Ang mga palatandaan na lumilitaw ay ganap na dahil sa cellular na lokasyon ng mutated gene. Sa ilang mga pasyente, ang diagnosis ay nakumpirma sa maagang pagkabata, dahil may mga halatang anomalya sa pag-unlad ng mga buto at mayroong isang pagtaas ng gawain ng mga glandula ng endocrine (halimbawa, ang regla ay maaaring magsimula sa pagkabata - sporadic menorrhagia).
Leukoplakia - mga sanhi sa mga bata at matatanda, pagsusuri, lokalisasyon at mga paraan ng paggamot
Impeksyon sa Enterovirus - sanhi ng ahente at mga ruta ng paghahatid, sintomas, pagsusuri, paraan ng paggamot at pag-iwas
Dressler's syndrome - mga sanhi pagkatapos ng atake sa puso, mga sintomas, pagsusuri, mga paraan ng paggamot at pag-iwas
Ang sakit na Albright ay nailalarawan sa pamamagitan ng tatlong pangunahing tampok, sa pagkakaroon ng dalawa sa tatlo, ang diagnosis na ito ay maaari nang gawin:
- Precocious puberty. Ang sintomas ay maaaring mahayag nang buo o hindi kumpleto. Halimbawa, ang regla ay nagsisimula nang maaga, ang mga ito ay sagana, masakit, ngunit sa isang hindi kumpletong anyo ng isang sintomas, maaaring wala sila. Sa pagkabata, lumilitaw ang pangalawang sekswal na mga katangian, tulad ng pamamaga at pag-unlad ng mga glandula ng mammary, kadalasang lumilitaw ang mga cyst sa mga ovary. Ang mga lalaki ay lumalaki ang pubic hair at isang titi.
- Fibrous osteodysplasia. Ang paglaki ng skeleton, maturity nito, at ossification ay higit na nauuna sa mga karaniwang indicator ng pag-unlad. Ang paglabag sa tisyu ng buto ay humahantong sa kurbada ng mga buto, ang kanilang pagpapapangit, kawalaan ng simetrya ay katangian. Ang pinakakaraniwang sintomas ay pagkapilay, kurbada ng gulugod. Ang paglaki ay bumagal, na kapansin-pansin na sa pagkabata, pagkatapos ay ang maikling tangkad ay nagsisimula na sumama sa sobrang timbang. Sa maraming mga pasyente, ang hindi pantay na paglaki ng bungo ay nangyayari, ang panlabas na pagpapapangit, ang mga nakaumbok na mata ay nangyayari. Sa edad ng pagdadalaga, bumabagal ang mga pagbabago sa buto, bumababa ang bilang ng mga bali.
- Mga pagbabago sa balat. Ito ay nagpapakita ng sarili sa hyperpigmentation. Ang balat sa katawan ay natatakpan ng malalaking dilaw-kayumanggi na mga spot na may hindi pantay na mga gilid. Ang mga lugar ng pigmentation ay maaaring matatagpuan sa apektadong bahagi sa rehiyon ng lumbar, sa hita, itaas na likod, puwit, dibdib. Sa isang bata, lumilitaw ang mga light brown spot, bilang panuntunan, kaagad pagkatapos ng kapanganakan.
Ang Albright's syndrome ay ipinakita sa pamamagitan ng mga palatandaan ng pinsala sa tissue ng buto, balat at mga sintomas ng mga endocrine disorder.
- Ang mga pasyente ay bumuo ng fibrous osteodysplasia, na ipinakita sa pamamagitan ng iba't ibang mga deformidad at skeletal curvature. Kadalasan, ang mga tubular bone sa isang paa ay apektado, na humahantong sa pagkapilay at spinal deformity. Nasa murang edad, ang pagbuo ng balangkas ay nabalisa. Sa mga pasyente, ang pagpapapangit ng panga at bungo ay ipinahayag: ang mga buto ng mukha ay lumapot, nagiging malawak, magaspang at malaki. Ito ay kung paano nabuo ang mga panlabas na deformidad: isang asymmetric na mukha at isang panig na exophthalmos. Pagkatapos ay lilitaw ang mga sintomas ng arthritis. Ang Fibrocystic dysplasia ay isang focal replacement ng cortical bone na may proliferating fibroblasts. Ang pagbuo ng mga cyst ay humahantong sa pagpapapangit ng buto at ang paglitaw ng mga bali. Ang pinakakaraniwang uri ng pagpapapangit ng balakang ay ang baluktot ng pastol.
- Ang mga lugar ng hyperpigmentation ay lumilitaw sa balat ng mga pasyente, na kung saan ay mga kumpol ng hindi regular na hugis na mga spot na may hindi pantay na mga gilid. Ang kulay ng mga spot ay madalas na nag-iiba mula sa madilaw-dilaw hanggang madilim na kayumanggi. Ang mga sugat ay naisalokal sa mga binti, hita, katawan. Ang mga spot ay sapat na malaki at may makinis na ibabaw. Ang mga ito ay naroroon sa balat mula sa kapanganakan o kumalat habang lumalaki ang bata. Ang epidermis ay hindi nagbabago. Ang kanilang intensive growth ay nangangailangan ng histological examination at surgical intervention.
- Ang mga endocrine disorder ay sanhi ng dysfunction ng mga istruktura ng hypothalamic-pituitary region at mga endocrine gland na matatagpuan sa periphery. Ang pagbibinata ay nagsisimula sa anim na buwang gulang na mga bata at nagtatapos nang maaga sa pitong taong gulang. Sa mga pasyente, nagsisimula ang regla, nabuo ang mga glandula ng mammary, lumilitaw ang pangalawang sekswal na mga katangian - paglaki ng dibdib at ang hitsura ng pubic hair. Sa panahon ng pagsusuri sa ultrasound ng mga ovary, ang malalaking follicular cyst ay matatagpuan sa kanila. Kasabay nito, ang mga ovary mismo ay nananatiling normal na laki.
- Ang mga pasyente ay nakakaranas ng tonic convulsions na nangyayari sa kanilang sarili o sa ilalim ng impluwensya ng endogenous at exogenous stimuli - stress, hypo- o hyperthermia, labis na overexertion.
- Ang mga calcification sa balat at malambot na mga tisyu ay kadalasang nagiging inflamed at ulceration, na nagiging sanhi ng kakulangan sa ginhawa sa mga pasyente.
- Mayroong osteochondromas, chondromatosis, subperiosteal resorption ng maliliit na buto ng mga daliri.
- Ang mga karaniwang panlabas na palatandaan ay maikli ang tangkad, hugis-buwan na mukha, sobra sa timbang, maiikling daliri.
- Ang labis na phosphorus ay nakakagambala sa pagsipsip ng magnesium, na nagiging sanhi ng migraines, arrhythmias, at pananakit ng likod.
- Ang pagsasaayos at kadaliang kumilos ng mga kasukasuan ay nabalisa, nabuo ang mga huwad na kasukasuan.
- Ang iba pang mga palatandaan ng sindrom ay kinabibilangan ng: pagsusuka, hematuria, pag-ulap ng lens, bahagyang kawalan ng enamel ng ngipin.
- Ang mga sakit sa neurological ay sanhi ng pagkasayang ng optic nerve, pagkawala ng pandinig at paningin.
- Naantala ang pag-unlad ng psychomotor.
Maglaan ng atypical o asymptomatic form ng pseudohypoparathyroidism, kung saan ang dami ng calcium at phosphorus sa dugo ay nananatiling normal, walang mga convulsion at osteomalacia.
Mga sanhi ng Albright Syndrome
Ang Albright's syndrome ay isang namamana na patolohiya na may hindi malinaw na pinagmulan. Tinatawag din itong pseudohypoparathyroidism. Ito ay nailalarawan sa pamamagitan ng pinsala sa tissue ng buto. Ang sakit ay nagpapakita ng sarili pangunahin sa babaeng kalahati ng populasyon, bagaman ito ay nangyayari rin sa mga lalaki. Halos lahat ng mga pasyente ay may mga paglihis sa mental at pisikal na pag-unlad.
Ang mga buto ng tao ay nagsisimulang masira sa ilalim ng impluwensya ng mga metabolic na proseso ng posporus at kaltsyum. Nagsisimula ang paglabas ng parathyroid hormone ng parathyroid gland.
Ang pangunahing dahilan para sa naturang patolohiya bilang Forbes-Albright syndrome ay ang namamana na pagtutol ng mga tisyu ng katawan sa impluwensya ng parathyroid hormone. Kung ang isang tao ay walang sakit, kung gayon ang hormone ay kumikilos sa pamamagitan ng isang tiyak na sangkap. Kapag ang sakit ay nagpapakita mismo, ang komposisyon ng mga sangkap na ito ay nagbabago sa antas ng genetic. Kaya, ang mga peripheral tissue ay nagiging insensitive sa parathyroid hormone.
Mga pangunahing pagpapakita
Ang lahat ng mga pagpapakita ng sakit na ito ay maaaring nahahati sa maraming uri. Halimbawa, ang mga pagpapakita ng endocrine ay kinabibilangan ng:
- Maikling tangkad.
- Obesity.
- Nakataas na asukal sa dugo.
- Lunar na mukha.
- Oligomenorrhea.
Sa ilang mga kaso, ang pseudohypoparathyroidism ay pinagsama sa acromegaly, Cushing's syndrome, gynecomastitis.
Ang mga pagpapakita ng osteoarticular ng patolohiya ay ipinahayag sa:
- Pagpapaikli ng mga daliri.
- Exostoses.
- Dyschondroplasia.
- Naantala ang pagngingipin.
- Hypoplasia ng enamel ng ngipin.
- Subcutaneous ossifications.
Ang mga sintomas ng pseudohypoparathyroidism ay ipinahayag din bilang paglabag sa nervous system. Kasabay nito, ang mga bata mula sa kapanganakan ay maaaring makaranas ng mga kombulsyon, pati na rin ang isang binibigkas na mental retardation.
Sa iba pang mga sintomas, ang urolithiasis, ang pagkakaroon ng dugo sa ihi, pagsusuka, at mga katarata ay madalas na nabanggit.
Sa bahagi ng mga endocrine disorder, nangyayari ang mga dysfunction ng pituitary gland at peripheral endocrine glands.

Ang pinakakaraniwang mga karamdaman ng sekswal na pag-unlad, na kung saan ay nailalarawan sa pamamagitan ng maagang pagkahinog. Ang napaaga na sekswal na pag-unlad ay bubuo, simula sa anim na buwan hanggang pitong taong gulang, depende sa mga indibidwal na katangian ng organismo. Ang patolohiya na ito ay ipinahayag ng buong anyo at hindi kumpleto.
Ang buong hitsura ay minarkahan ng pagkakaroon ng dalawang palatandaan: ang maagang pagsisimula ng regla at ang pag-unlad ng mga glandula ng mammary. Sa panahon ng hindi kumpleto, ang pagpapakita ng pangalawang sekswal na mga katangian na walang regla ay katangian. Kaya, ang pinabilis na paglaki sa taas ay maaaring lumitaw, gayunpaman, sa edad ng reproductive, ang mga batang babae ay hindi naiiba sa mga kababaihan na may normal na pag-unlad.
Sa bahagi ng mga endocrine disorder, nangyayari ang mga dysfunction ng pituitary gland at peripheral endocrine glands.
Kung ang buong anyo ay pumasa sa pinabilis na pag-unlad, kung gayon ang hindi kumpletong anyo ng pag-unlad ay pinahaba ng maraming beses.
Ang Albright's syndrome ay naiiba sa tunay na masyadong maagang pag-unlad sa na, na may hormonal dysfunctions, sekswal na paglaki ng buhok ay hindi nangyayari, at isang bahagyang acceleration ng skeletal differentiation ay nabanggit.
Ang mga malalaking follicular cyst ay nabuo sa mga ovary, ngunit ang mga ovary mismo ay hindi tumataas. Ang mga gonadotropic hormone ay kadalasang nananatiling hindi nagbabago.
Mayroon ding isang bilang ng mga endocrine pathologies, kabilang ang hyperparathyroidism, hyperthyroidism, acromegaly, Itsenko-Cushing's syndrome, rickets.
Posible ang fibrous osteodysplasia, na ipinakikita ng pagbabago sa ossification, iba't ibang deformities, at skeletal curvature. Karaniwan para sa Albright syndrome na magdulot ng pinsala sa mga tubular na buto sa isang bahagi ng balangkas, na humahantong sa pagkapilay at deformity ng spinal column - ang kundisyong ito ay makikita sa maraming larawan sa Internet.
Ang mga rate ng paglaki at pagbuo ng kalansay, sa maraming mga kaso, ay naiiba na sa murang edad. Sa isang mas malaking porsyento ng mga pasyente, mayroong isang pagpapapangit ng mga buto ng panga at bungo, bilang isang resulta kung saan mayroong isang abnormal na paglaki ng mukha, na, dahil sa mga pagbabago sa hormonal, ay humahantong sa acromegaly (ang sakit ay nailalarawan sa pamamagitan ng pampalapot ng ang mga kamay at facial area ng bungo, at sila rin ay nagiging masyadong malawak) at iba pang mga pathologies na nagpapakita ng kanilang sarili sa mga panlabas na deformidad.
Kaya, sa panahon ng pananaliksik, sa mga bata na nasuri na may acromegaly, ang hitsura ng masyadong magaspang na mga tampok ng mukha ay nabanggit, ang mas mababang at itaas na mga paa ay pinalaki nang malaki, bilang karagdagan sa kasalukuyang sakit, lumitaw ang mga sintomas ng arthritis.
Sa bahagi ng balat, ang hyperpigmentation ay nabanggit, na lumilitaw sa anyo ng isang kumpol ng mga spot o isang patag na hindi regular na hugis na may hindi pantay na mga gilid. Ang kulay ng mga spot ay maaaring madilaw-dilaw o kayumanggi. Ang lokasyon ng hyperpigmentation ay pangunahin sa mga binti, sa hita o sa puno ng kahoy.
Mga kadahilanan na nag-aambag sa pag-unlad ng sindrom:
- congenital anomalya ng hypothalamus,
- psychogenic na mga kadahilanan - stress, salungatan, neuroses,
- endocrinopathy,
- skull base hyperostosis,
- napaaga na pagtatago ng gonadotropins.
Ang Albright's syndrome ay resulta ng malfunction ng mga glandula ng parathyroid. Ang kanilang pangunahing pag-andar ay ang regulasyon ng metabolismo ng phosphorus-calcium. Sa patolohiya na ito, ang mga glandula ay hindi maganda ang reaksyon sa mga panginginig ng boses ng mga elementong ito o hindi nila nakikita ang mga ito.
Ang genetically determined resistance ng mga structural elements ng kidney at skeleton sa parathyroid hormone ay dahil sa pinsala sa cytoreceptors o dysfunction ng adenylate cyclase. Ang mutation ng mga partikular na gene ay humahantong sa pagsugpo sa produksyon ng cAMP, na kasangkot sa metabolismo na nangyayari sa ilalim ng impluwensya ng parathyroid hormone.
Ang mga sanhi ng kakulangan sa cAMP ay iba-iba:
- pinsala sa mga protina na nagbubuklod ng nucleotide ng mga lamad ng cell,
- depekto ng epithelial cytoreceptor,
- malfunction ng adenylate cyclase.
Ginagawa ng mga pathological na pagbabagong ito ang mga peripheral tissue na buo sa parathyroid hormone. Ang mga target na organo ay nawawala ang kanilang sensitivity sa parathyroid hormone, na humahantong sa isang pagtaas sa antas ng posporus sa dugo at pagbaba sa konsentrasyon ng calcium.
Ang buto porosity at cyst formation ay mga katangiang pagbabago na makikita sa sindrom na ito. Ang kaltsyum ay hugasan sa labas ng mga buto at idineposito sa anyo ng mga calcification sa subcutaneous fat, kalamnan, panloob na organo, endothelium ng mga arterial vessel. Sa paglipas ng panahon, ang bone tissue ay napapalitan ng fibrous tissue. Ang mga skeletal anomalya ay nakakapinsala sa paggalaw ng mga pasyente at nagdudulot ng matinding pananakit.
Ang pagbaba ng bitamina D sa dugo ay humahantong sa isang paglabag sa pagsipsip ng calcium sa bituka. Ang hypocalcemia ay nagpapasigla ng mas malaking pagpapalabas ng parathyroid hormone sa dugo at nadagdagan ang pag-leaching ng calcium mula sa mga buto, na humahantong sa pagbuo ng malubhang osteomalacia. Ang mga sakit sa buto sa sakit na ito ay itinuturing na pinakamalubha.
Ang mga brown pigment spot na may hindi pantay na mga gilid ay lumilitaw sa balat ng mga pasyente. Ang mga endocrine disorder sa sakit na ito ay kinakatawan ng maagang pagdadalaga at ang hitsura ng pangalawang sekswal na katangian.
Ang isang kahila-hilakbot na diagnosis na tinatawag na Albright syndrome ay may hindi malinaw na etiology.
Ang patolohiya ng genetic na pinagmulan ay ipinakita sa pamamagitan ng pagtaas ng pigmentation ng balat at mga pagbabago sa hormonal system na may maagang pag-unlad ng sekswal. Ang isang bihirang sakit ay nakakaapekto sa buong katawan ng tao.
Maraming mga pasyente ang nagdurusa hindi lamang mula sa pisikal na pagkaantala, ang mga pasyente na may Albright's disease ay nailalarawan din ng kapansanan sa pag-iisip.
Ang pseudohypoparathyroidism ay tinatawag ding patolohiya, na pinangalanan sa American scientist na si Fuller Albright, na natuklasan ang sakit na ito.
Ang dalas ng sakit ay nangyayari sa buong mundo bilang isang kaso bawat daang libo o isang milyong tao.
Ang pagtatatag ng diagnosis ay mahirap, at sa kadahilanang ito, ang sakit na Albright ay maaaring maling makilala, na nagpapahirap sa tumpak na pagtukoy ng data sa pagkalat nito.
Ang McCune-Albright syndrome (isa pang kasingkahulugan para sa patolohiya) ay bubuo dahil sa genetic mutations na nagsisimulang mangyari sa katawan nang hindi sinasadya, nang walang dahilan.
Ang parathyroid gland ng katawan, na kumokontrol sa antas ng calcium at phosphorus, ay naglalabas ng parathyroid hormone. Sa isang tao na nagkasakit ng tulad ng isang sindrom, ang resistensya ng tisyu sa hormone na ito ay may kapansanan.
Bilang isang resulta, ang sistema ng kalansay ng tao ay nagsisimulang masira sa ilalim ng impluwensya ng mga resulta ng mga pagbabago sa metabolismo ng calcium at phosphorus.
Ang sindrom ay nailalarawan sa pamamagitan ng hitsura sa katawan ng mga tiyak na mga spot, fibrous dysplasia at iba't ibang mga endocrinopathies. Kabilang sa huli, ang pinakakaraniwan ay maagang pagbibinata.
Ang ilang mga geneticist ay nagtatalo tungkol sa napakaraming dalas ng mga pasyente sa mga batang babae, dahil ang katangian ng sintomas ng maagang pagdadalaga sa mga batang babae ay nabanggit halos 10 beses na mas madalas. Ang iba pang mga pagpapakita ng sindrom ay nangyayari sa parehong kasarian halos pantay.
Ang etiology ng sindrom ay hindi alam.
Ang mga neurogenic na impluwensya at endocrine mutations ay ipinapalagay, na sanhi ng congenital abnormalities sa hypothalamic region sa panahon ng embryonic development at maagang produksyon ng gonadotropic hormone.
Naniniwala ang mga siyentipiko na ang pag-unlad ng McCune-Albright syndrome ay sanhi ng kakulangan ng tugon ng mga selula ng bato at buto sa pagkilos ng mga parathyroid hormones.
Ang antas ng kaltsyum at posporus sa sistema ng sirkulasyon ay kinokontrol ng mga glandula ng parathyroid, ngunit sa kondisyong ito ng pathological, ang kanilang buo (walang malasakit) na tugon sa mga pagbabago sa mga elementong ito ay humahantong sa hyperparathyroidism (labis na produksyon ng parathyroid hormone), mga pagbabago sa istraktura ng buto .
Sa McCune-Albright syndrome, ang pagkabigo ay sanhi ng mutation ng gene.
Ang paglabag ay maaaring mangyari kahit saan sa metabolic chain: sa ilang mga pasyente, ang receptor ay nasira, sa isang bilang ng mga pasyente, ang mga protina ng cell lamad ay sumailalim sa isang pagbabago, sa iba, mayroong hindi sapat na produksyon ng cAMP.
Ang genetic na kondisyon na nagkokonekta sa mga kasong ito ay hindi minana: ang mutation ay nangyayari pagkatapos ng fertilization.
Mga sintomas
Ang mga palatandaan na lumilitaw ay ganap na dahil sa cellular na lokasyon ng mutated gene.
Sa ilang mga pasyente, ang diagnosis ay nakumpirma sa maagang pagkabata, dahil may mga halatang anomalya sa pag-unlad ng mga buto at mayroong isang pagtaas ng gawain ng mga glandula ng endocrine (halimbawa, ang regla ay maaaring magsimula sa pagkabata - sporadic menorrhagia).
Sa ibang mga pasyente, lumilitaw ang mga sintomas sa ibang pagkakataon. Ang pangunahing mga palatandaan ng pathological ay sinamahan ng isang pangkalahatang lag sa pag-unlad ng tao - intelektwal at pisikal.
Mga diagnostic
Ang sakit na Albright ay mahirap masuri sa isang sanggol, at sa edad na 6-10 taon, ang mga sintomas ay binibigkas na. Mayroong maraming mga paglihis sa pag-unlad ng skeletal system, ang kaltsyum sa dugo ay nabawasan, ang posporus at parathyroid hormone ay nadagdagan.
Mga sintomas
Mayroong tatlong pangunahing sintomas ng sakit. Sa pagkakaroon ng kahit dalawa sa tatlo, ang gayong pagsusuri ay maaaring gawin.
- Napakaaga ng pagdadalaga bilang resulta ng napakataas na background ng hormonal. Maaaring magsimula ang regla sa mga batang babae sa mga unang buwan pagkatapos ng kapanganakan. Sa mga batang lalaki na nagdurusa sa Albright syndrome, ang maagang pagdadalaga ay napakabihirang naobserbahan. Ang isang pagbabago sa hormonal background ay nangyayari dahil sa hindi tamang paggana ng adrenal glands, pituitary gland at thyroid gland. Maaaring lumitaw ang sintomas na ito sa buo o hindi kumpletong anyo. Sa buong anyo ng regla, nagsisimula sila nang maaga, kadalasan sila ay sagana at masakit. Sa pagkabata, ang sindrom na ito ay nagpapakita rin ng pangalawang sekswal na mga katangian, halimbawa, pamamaga ng mga glandula ng mammary. Sa hindi kumpletong pagpapakita ng regla ay hindi mangyayari. Kadalasan, na may Albright syndrome, ang mga batang babae ay nagkakaroon ng mga cyst sa mga ovary. Sa mga batang lalaki na may maagang pagdadalaga, mayroong pagtaas sa ari ng lalaki at pubic hair.
- Fibrous osteodysplasia. Ang sakit na ito ay nagpapahiwatig ng isang paglabag sa tissue ng buto, humahantong sa kurbada at pagpapapangit ng mga buto. Nailalarawan sa pamamagitan ng kawalaan ng simetrya. Ang pinakakaraniwang mga palatandaan ay pagkapilay at kurbada ng gulugod. Kadalasan, ang mga tubular bone ay sumasailalim sa mga pagbabago. Ang paglaki ng balangkas ay nagpapabagal, ito ay kapansin-pansin kahit sa mga bata. Maraming biktima ng McCune syndrome ang may hindi pantay na paglaki ng mga buto ng bungo, na humahantong sa panlabas na deformity. Sa isang pagbabago sa mukha, posible ang pagpapakita ng mga nakaumbok na mata. Matapos maabot ang edad ng pagdadalaga, bumabagal ang mga pagbabago sa buto at bumababa ang dalas ng mga bali.
- Mga pagbabago sa balat. Naipapakita sa pagtaas ng pigmentation. Ang balat sa puno ng kahoy at hita ay natatakpan ng dilaw-kayumanggi na mga spot na may hindi pantay na mga gilid. Ang pigmentation ay makikita sa lower back, upper back, buttocks, at chest. Lumilitaw kaagad ang mga spot pagkatapos ng kapanganakan ng bata.
Ang Albright syndrome ay isang namamana na sakit na kinabibilangan ng isang kumplikadong mga pathologies ng endocrine system, balat at musculoskeletal system. Minsan ang sakit na ito ay tinatawag na pseudohypoparathyroidism, hereditary osteodystrophy, o "Javan chicken syndrome."
Noong 1942, ang patolohiya na ito ay unang naitala ng Amerikanong manggagamot na si Fuller Albright, na nag-aral ng endocrine system, lalo na ang metabolismo ng calcium.
Ang Albright's syndrome ay napakabihirang, na may tinatayang saklaw na 7.9 bawat 1 milyong tao, habang ang patolohiya ay mas karaniwan sa mga babae.
Ang anumang namamana na patolohiya ay sanhi ng mga negatibong mutasyon sa mga gene, na nagiging sanhi ng mga karamdaman sa pag-unlad ng anumang sistema ng katawan na nauugnay sa paggana ng mga gene na ito.
Ang sakit na Albright ay nailalarawan sa pamamagitan ng mga mutasyon sa gene ng GNAS1, na nag-encode ng isang espesyal na protina ng Gs - alpha. Ang protina na ito ay direktang nagbubuklod sa mga receptor ng parathyroid hormone (PTH).
Ang pamana ng Albright osteodystrophy ay nauugnay sa X chromosome, bilang ebidensya ng kakulangan ng mana mula sa ama sa anak na lalaki at ang mataas na saklaw ng sakit sa mga babae.
Ang pagbawas sa aktibidad ng GS-alpha protein ay humahantong sa katotohanan na ang mga tisyu ng katawan ay nagiging insensitive sa parathyroid hormone. Bilang karagdagan, ang synthesis ng cAMP, isang molekula na gumaganap bilang isang tagapamagitan para sa pagpapalaganap ng mga signal ng ilang mga hormone, ay nagambala sa mga selula.
Mga sintomas
Brachydactyly sa Albright's syndrome Ang sakit ay ipinapakita sa pamamagitan ng maikling tangkad Hugis-buwan na hugis ng mukha
Depende sa sanhi ng patolohiya, ang ilang mga uri ng sakit ay nakikilala.
Ang pseudohypoparathyroidism, na kung saan ay nailalarawan sa pamamagitan ng patolohiya ng mga cellular receptor na nagbubuklod sa parathormone, ay nakahiwalay sa uri 1a na sakit. Ang isa pang uri ng sakit, 1b, ay nailalarawan sa pamamagitan ng isang paglabag sa mga protina na nagbubuklod sa mga receptor at enzyme sa isa't isa.
Ang pseudohypoparathyroidism type 2 ay nailalarawan sa pamamagitan ng isang matinding kakulangan ng cAMP.
Anuman ang uri ng sakit, ang sakit na Albright ay nailalarawan sa pamamagitan ng ilang mga pangkalahatang sintomas. Dahil sa isang paglabag sa pag-unlad ng endocrine system, ang mga pasyente ay maaaring makaranas ng pagtaas sa mga glandula ng parathyroid. Ang katawan ay maaaring bumuo ng hindi katimbang dahil sa pagpapaikli ng mas mababang mga paa't kamay, na humahantong sa kapansin-pansing dwarfism (tingnan ang larawan sa itaas).
Ang mga daliri ay maaari ding maging maikli, na tinatawag na brachydactyly. Ang hugis ng mukha ay "hugis-buwan". Maaaring mangyari ang labis na katabaan at osteoporosis. Dahil sa kapansanan sa pag-unlad ng musculoskeletal system, ang spinal deformity, ang paglitaw ng mga maling joints at kawalan ng kadaliang kumilos sa mga lugar kung saan dapat ito ay nasa natural na anatomical na paraan ay maaaring mabilis na umunlad.
Kadalasan, ang mga pasyente na may Albright's disease ay nagkakaroon ng Itsenko-Cushing's syndrome.
Dahil sa hindi tamang pag-unlad ng tissue ng buto, ang mga problema ay lumitaw sa mga ngipin na nauugnay sa mabilis na pinsala sa enamel. Dahil sa hypocalcemia, may mataas na panganib na magkaroon ng katarata.
Ang Albright syndrome ay nailalarawan din sa pamamagitan ng soft tissue calcification dahil sa calcium na inilabas mula sa mga buto.
Ang mga akumulasyon ng calcium ay maaaring mabuo sa mga bato, iba't ibang mga kalamnan, sa mga dingding ng mga daluyan ng dugo at sa myocardium, na maaaring humantong sa mga komplikasyon sa puso.
Sa pangkalahatan, ang mga calcification, kahit na nasa subcutaneous tissue, ay maaaring makaapekto sa mga tisyu sa kanilang paligid, na nagiging sanhi ng maliliit, ngunit hindi komportable na mga ulser na mabuo.
Bilang karagdagan sa mga pagbabago sa itaas, lumilitaw ang mga kulay ng kape na may iba't ibang laki sa balat ng pasyente (tingnan ang larawan sa ibaba).
Paglabag sa pigmentation ng balat (lumilitaw ang mga brown spot sa katawan na katulad ng isang nunal)
Ang isang hiwalay na punto sa listahan ng mga neurological disorder ay tonic convulsions. Maaari silang mangyari pareho sa kanilang sarili at dahil sa pagkilos ng anumang mga irritant, tulad ng stress, nakakapagod na pisikal na trabaho, biglaang pagbabago sa temperatura, atbp.
Mga diagnostic
Dahil ang pseudohypoparathyroidism ay isang namamana na sakit, kung may mga taong may katulad na patolohiya sa pedigree, lalo na malapit na kamag-anak, kinakailangan na kumunsulta sa isang geneticist upang masuri ang panganib ng pagkakaroon ng isang may sakit na bata.

Medyo mahirap na agad na masuri ang Albright's syndrome sa isang bagong panganak, ngunit sa pagkabata (5-10 taon), posible na ito, dahil lumilitaw ang lahat ng mga sintomas na katangian ng sakit.
Kinakailangan din na magsagawa ng isang bilang ng mga pagsusuri, sa partikular, isang pagsusuri sa dugo upang matukoy ang antas ng calcium, phosphorus at parathyroid hormone. Maaaring matukoy ng urinalysis ang dami ng phosphorus at cAMP.
Mayroong ilang mga espesyal na pagsusuri sa diagnostic na naglalayong pag-aralan ang pagiging sensitibo ng tissue sa parathyroid hormone.
Ang mga diagnostic ng X-ray ay maaaring magpahiwatig ng mga pagbabago sa balangkas at Ang kumbinasyon ng mga resulta ng pagsusuri, pisikal at mental na mga sintomas ay magsasaad kung anong uri ng sakit ito at kung ito ay Albright's syndrome, dahil ang mga katulad na sintomas ay maaaring maobserbahan sa mga namamana na sakit tulad ng Shereshevsky- Turner syndrome.
Ang maagang pagsusuri ng pseudohypoparathyroidism ay napakahalaga, na humahantong sa isang mabilis na pagsisimula ng paggamot. Kung mas mabilis na sinimulan ang kinakailangang paggamot, mas mababa ang panganib ng mga anomalya ng pag-unlad ng kaisipan. Kadalasan ang mga pasyente na may Albright syndrome ay sinusunod ng ilang mga espesyalista:
- gynecologist o urologist;
- radiologist;
- endocrinologist;
- orthopedist;
- nutrisyunista, atbp.,
Ito ay dahil sa mga kumplikadong sintomas ng patolohiya.
Halos palaging, ang mga pasyente ay inireseta ng iba't ibang mga paghahanda ng calcium, dahil, hindi katulad ng mga pasyente na may totoong hypoparathyroidism, ang mga iniksyon ng parathyroid hormone ay hindi humahantong sa nais na epekto.
Napakahalaga na piliin ang tamang dosis upang mapanatili ang normal na homeostasis.
Para sa isang mas kumpletong pagsipsip ng calcium, kasama ang mga paghahanda batay dito, ang mga bitamina complex ay inireseta, lalo na ang mga naglalaman ng bitamina D, halimbawa, tulad ng:
- Oksidevit;
- Calcitriol;
- Dihydrotachysterol.
Bilang karagdagan sa pag-inom ng gamot, ang mga pasyente ay kailangang sumunod sa isang mahigpit na diyeta, batay din sa mga pagkaing mayaman sa calcium.
Ang kabaligtaran na epekto ay hindi dapat pahintulutan - isang labis na antas ng kaltsyum sa itaas ng pamantayan, dahil maaaring mangyari ang hypercalcemia, na sinamahan ng pagkasira, pananakit ng ulo, at mga karamdaman ng gastrointestinal tract.
Sa kasong ito, kinakailangan na ganap na iwanan ang mga paghahanda ng calcium at, una sa lahat, pagalingin ang hypercalcemia.

Kung ang sakit ay sinamahan ng mga makabuluhang kaguluhan ng endocrine system, kung gayon ang pangangailangan para sa therapy ng hormone ay maaaring kailanganin. Ang Tamoxifen, isang gamot na humaharang sa mga receptor ng estrogen, ay kadalasang ginagamit para sa therapy sa hormone.
Ang antas ng kaltsyum ay sinusuri bawat linggo upang masuri kung kinakailangan upang ayusin ang kurso ng paggamot o hindi. Kasunod nito, ang antas ng calcium ay sinusuri isang beses sa isang buwan, at pagkatapos, kung ang mga kanais-nais na resulta ay nakamit, isang beses sa isang quarter.
Sa mga convulsion, ang isang 10% na solusyon ng chloride o calcium gluconate ay ibinibigay sa intravenously. Napakahalaga na mag-inject ng solusyon nang dahan-dahan, dahil may panganib ng pagkalasing. Inirerekomenda na ulitin ang intravenous injection 2-3 beses sa isang araw.
Kung ang anumang partikular at tiyak na mga karamdaman ay sinusunod, pagkatapos ay ginagamot sila ng naaangkop na mga espesyalista, halimbawa, mga katarata - isang oculist, at mga sakit sa ngipin - isang dentista.
Sa mga matatanda, ang mga pathological na pagbabago sa pag-unlad ng tissue ng buto.
Operasyon
Ang operasyon para sa Albright syndrome ay ginagawa kapag ang mga abnormalidad ng buto ay maaaring makapinsala sa nerve.
Mga diagnostic
Etiopathogenetic na mga kadahilanan
Ang pseudohypoparathyroidism ay batay sa namamana na pagtutol ng mga tisyu ng katawan sa impluwensya ng parathyroid hormone. Sa ilalim ng normal na mga kondisyon, ang huli ay kumikilos sa pamamagitan ng isang espesyal na tagapamagitan - cyclic adenosine monophosphate (cAMP). Siya ang nagtuturo sa cell na sumunod sa regulatory signal ng parathormone. Sa sakit na Albright, nagbabago ang istraktura ng tagapamagitan sa antas ng genetic.
Ang sakit na Albright ay ipinapasa sa mga susunod na henerasyon sa isang kawili-wiling pattern na tinatawag na X-linked dominant inheritance: ang patolohiya mula sa ina ay pumasa sa mga anak na babae at lalaki sa 50% ng mga kaso. Mula sa ama na may parehong posibilidad, ang mga anak na babae lamang ang nagmamana ng may sira na gene. Mayroong mga kaso ng pamilya.
Ang gene para sa Albright syndrome ay matatagpuan sa sex X chromosome
Kung wala ang nagre-regulate na pagkilos ng parathyroid hormone, ang metabolismo ng calcium ay makabuluhang naililipat patungo sa paggamit sa tissue ng buto. Kasabay nito, ang kakulangan nito ay sinusunod sa dugo, na humahantong sa mga naturang phenomena:
- kalamnan cramps;
- paglabag sa pagbuo ng mga electrical impulses sa myocardium at isang pagbabago sa rate ng puso;
- nadagdagan ang produksyon ng parathyroid hormone at isang pagtaas sa mga glandula ng parathyroid.
Ang labis na parathyroid hormone, naman, ay humahantong sa pagtaas ng antas ng posporus sa dugo.
Ang thyroid at parathyroid glands ay ang pangunahing regulators ng metabolismo ng calcium at phosphorus sa katawan.
Ang genetic na patolohiya na ito ay nailalarawan sa pamamagitan ng isang autosomal na nangingibabaw na mode ng mana.
Ang pangunahing dahilan ay isang depekto ng kapanganakan na ginagawang hindi sensitibo ang mga peripheral tissue sa parathyroid hormone.
Depende sa kung paano at kung saan nasira ang metabolic chain, dalawang uri ng sindrom ay nakikilala.
Paggamot ng sakit
Ang mga karaniwang nakikitang palatandaan ay kinabibilangan ng:
- mababang paglago;
- sobra sa timbang;
- mataas na antas ng glucose;
- mukha ng buwan;
- mga karamdaman sa pagdadalaga.
Ang mga sumusunod na sintomas ay maaari ding lumitaw:
- acromegaly;
- mga sakit sa autoimmune;
- gynecomastia;
- Itsenko-Cushing's disease, atbp.
Ang mga pasyente ay madalas na nagreklamo ng dugo sa ihi, patuloy na pagsusuka, pagbabago sa ngipin, kombulsyon. Sa pagsusuri, makikita ang subcutaneous ossification, at maaaring lumitaw ang calcifications.
Kadalasan, ang mga pasyente ay pinaikli ang mga daliri, ang mga epiphyses ng tubular bones ay deformed, at maaaring may mga palatandaan ng isang lenticular cataract.
Halos palaging, ang mga naturang pasyente ay nahuhuli sa pag-unlad ng kaisipan.
Ang pseudohypoparathyroidism ay ginagamot nang komprehensibo.
Dahil ito ay isang genetic na patolohiya, ang therapy ay naglalayong alisin ang mga sintomas na lumitaw:
- Ang hormonal na paggamot ay inireseta. Makakatulong ito sa gawain ng endocrine system. Kinakailangan na maingat na subaybayan ang pagpapapangit ng mga buto ng bungo upang maiwasan ang pag-unlad ng patolohiya sa oras.
- Interbensyon sa pagpapatakbo. Ang pamamaraang ito ay ginagamit kapag ang pagpapapangit ng mga buto ng mukha ay maaaring humantong sa kapansanan sa pandinig at paningin.
- Magreseta ng mga painkiller na naglalaman ng bisphosphonates.
Para sa bawat pasyente na nagdurusa sa Albright's syndrome, ang paraan ng therapy ay pinili nang paisa-isa. Dapat na mahigpit na sundin ng pasyente ang lahat ng mga rekomendasyon at reseta ng doktor.
Ang pana-panahong pagsubaybay sa antas ng calcium sa katawan ng pasyente ay isinasagawa. Sa paunang yugto ng paggamot, ang naturang pag-aaral ay isinasagawa minsan sa isang linggo. Unti-unti, ang dalas ng mga sukat ay nababawasan sa isang beses sa isang buwan. Ito ay isasagawa hanggang sa katapusan ng iniresetang paggamot.
Kung ang pseudohypoparathyroidism ay nasuri, kung gayon ang pasyente ay dapat sumunod sa isang mahigpit na diyeta. Binubuo ito sa kumpletong pagbubukod mula sa menu ng mga produkto kung saan naroroon ang posporus.
Ang isang namamana na sakit ay kadalasang nakakaapekto sa buong katawan, na humahantong sa maraming pagbabago. Ang mga palatandaan ng patolohiya sa kasong ito ay lubos na katulad sa mga hypoparathyroidism - isang kakulangan sa produksyon ng parathyroid hormone sa parathyroid glands.
Ang Albright syndrome ay isang genetic na sakit ng hindi kilalang etiology na nakakaapekto sa buong katawan ng tao. Ang patolohiya ay nangyayari medyo bihira at ipinahayag sa pamamagitan ng asymmetric hyperpigmentation ng balat, hormonal imbalance, polyostotic fibrous bone dysplasia. Ang karamdamang ito ay tumutukoy sa mga bihirang uri ng maagang pagdadalaga na umaasa sa gonadotroph.
Ang Albright's syndrome ay pangunahing bubuo sa mga kinatawan ng magandang kalahati ng sangkatauhan. Ito ay sanhi ng isang kusang mutation ng isang gene sa panahon ng embryogenesis at hindi minana. Ang diagnosis ay ginawa sa mga batang may edad na 5-10 taon batay sa mga palatandaan na katangian ng sakit na ito.
Sa unang pagkakataon, ang klinika ng sindrom ay inilarawan noong 1907 ng siyentipikong si Albright, kung saan nakuha niya ang kanyang pangalan. Napagmasdan ng siyentipiko ang maikli at pandak na mga batang babae na may isang bilog na mukha at isang maikling leeg. Napag-alaman na mayroon silang kalamnan, mga deformidad ng skeletal, bahagyang kawalan ng enamel ng ngipin, pagkaantala sa pag-iisip, at mga palatandaan ng labis na katabaan.
Ang endocrinopathy ay ipinakita sa maagang pagbibinata: ang hitsura ng regla, aktibong paglaki ng mga glandula ng mammary, ang hitsura ng pubic hair. Ipinaliwanag ni Albright ang pinagmulan ng patolohiya sa pamamagitan ng paglaban ng renal epithelium sa parathyroid hormone, na humahantong sa pinabilis na reabsorption ng phosphorus at pag-unlad ng hyperphosphatemia.
Kasabay nito, sa mga pasyente, ang mga glandula ng parathyroid ay may normal na istraktura, at walang mga pagbabago sa humoral.
Sa opisyal na gamot, ang sindrom ay tinatawag na pseudohypoparathyroidism. Ang mga paglabag sa metabolismo ng phosphorus-calcium ay humantong sa patolohiya ng skeletal system. Ang ganitong mga karamdaman ay madaling ipinaliwanag sa pamamagitan ng tissue resistance sa endogenous parathyroid hormone. Ang mga pasyente ay may lag sa psychophysical development. Kasabay nito, walang reaksyon sa pagpapakilala ng exogenous parathyroid hormone.
Ang mga pasyente na may Albright syndrome ay nangangailangan ng konsultasyon sa mga espesyalista sa larangan ng gynecology, endocrinology, radiation diagnostics, ophthalmology, traumatology at orthopedics.
Ang sakit na ito ay pangunahing nangyayari sa mga batang babae.
Lumilitaw ang mga karamdaman bilang resulta ng mutation ng gene sa embryonic stage of development, wala itong kakayahang magmana. Ang mga sintomas na lumilitaw ay ganap na nagmumula sa cellular na lokasyon kung saan pumapasok ang mutated gene.
Mga diagnostic
Ang kawalan ng pagtaas ng resistensya ng bato sa parathyroid hormone ay isang sapat na batayan para sa diagnosis ng Albright's disease.
Ang pagsasagawa ng biochemical blood test, na nagsiwalat ng hypophosphaturia (hindi sapat na paglabas ng phosphorus sa ihi), ay dinadagdagan ng x-ray na pagsusuri upang masubaybayan ang mga pagbabago sa mga buto at malambot na tisyu.
Ang mga kababaihan ay dapat sumailalim sa isang pagsusuri ng isang gynecologist, isang pagsusuri sa ultrasound ng pelvis at isang pagsusuri ng sex chromatin. Ang mga spot ay makikita sa kapanganakan, ngunit dahil sa kanilang maliit na sukat at pamumutla, sila ay madalas na hindi napapansin.
Pag-iwas
Ang pag-iwas ay ang mga sumusunod:
- ang mga taong may ganitong patolohiya ay dapat na regular na mag-ehersisyo upang palakasin ang kanilang mga kalamnan;
- kung ang isang babae ay gustong mabuntis, kailangan niyang kumunsulta sa isang gynecologist at isang geneticist.
Ang patolohiya na ito ay nasuri sa isang tao bawat isang milyong naninirahan sa planeta. Sa kasamaang palad, wala pang partikular na therapy. Mayroon lamang sintomas na paggamot. Ngunit kung ang pasyente ay nasa ilalim ng pangangasiwa ng isang doktor, mayroong isang pagkakataon para sa isang kanais-nais na kinalabasan.
Mga sintomas
- Dahil sa mataas na antas ng hormonal, nangyayari ang maagang pagdadalaga. Ang mga batang babae ay maaaring magsimula ng regla sa unang sampung buwan ng buhay. Ngunit ang sintomas na ito ay hindi nalalapat sa mga lalaki. Nagsisimula sila sa pagbibinata gaya ng dati. Ang hormonal background ay nagdaragdag kapag ang mga malfunctions ay nangyari sa gawain ng adrenal glands, thyroid gland at pituitary gland. Ang sintomas na ito ay maaaring bumuo sa dalawang anyo - hindi kumpleto at kumpleto. Kung ang patolohiya ay nasa buong anyo, kung gayon ang regla ay nagsisimula nang maaga. Gayunpaman, ang mga ito ay masakit at sagana. Lumilitaw din ang mga pangalawang sekswal na katangian, halimbawa, ang mga glandula ng mammary ay tumaas. Kung ang anyo ng patolohiya ay hindi kumpleto, pagkatapos ay hindi magsisimula ang regla. Kadalasan, ang sakit na Albright sa mga batang babae ay sinamahan ng pag-unlad ng mga cyst sa mga ovary. Ang maagang pagdadalaga sa mga lalaki ay nailalarawan sa pamamagitan ng pagpapalaki ng ari ng lalaki at ang hitsura ng pubic hair.
- Fibrous osteodysplasia. Ito ay isang patolohiya kung saan ang tissue ng buto ay deformed, dahil sa kung saan ang mga buto ng balangkas ay baluktot. Lumilitaw ito bilang kawalaan ng simetrya. Ang mga visual na sintomas ay kurbada ng gulugod at pagkapilay. Ang patolohiya na ito ay nakakaapekto sa pangunahing mga tubular bones. Bumagal ang paglaki ng tao. Ito ay makikita kahit sa mga bata. Ang mga buto ng bungo ay umuunlad nang hindi pantay, lumilitaw ang mga nakaumbok na mata. Kapag natapos na ang pagdadalaga, hihinto ang pagbabago ng buto.
- Pagbabago sa kulay ng balat. Ang malakas na pigmentation ay nagsisimulang lumitaw. Lumilitaw ang dilaw-kayumanggi na mga spot na may hindi malinaw na mga gilid sa mga hita at sa katawan mismo. Ang mga spot ay malinaw na nakikita sa ibabang likod, itaas na likod, dibdib at pigi. Sa Albright's syndrome, ang mga spot sa katawan ng sanggol ay lilitaw kaagad pagkatapos ng kapanganakan.
Ang sakit na pseudohypoparathyroidism ay nagpapakita ng mga sintomas ng isang tiyak na kalikasan.
Ang congenital hip dislocation ay isa sa mga pinakakaraniwang anomalya sa pag-unlad. Ang underdevelopment o dysplasia ng hip joint ay parehong unilateral at bilateral. Ang mga dahilan para sa pag-unlad ng patolohiya ay hindi lubos na nauunawaan, ngunit alam ng mga clinician ang isang malawak na hanay ng mga predisposing na kadahilanan na maaaring kumilos bilang isang provocateur ng sakit, mula sa isang genetic predisposition sa isang hindi sapat na pagbubuntis.
Osteochondropathy (nagkakasabay na sintomas: 2 sa 11)
Ang Osteochondropathy ay isang kolektibong konsepto na kinabibilangan ng mga sakit na nakakaapekto sa musculoskeletal system, laban sa background ng deformation at nekrosis ng apektadong segment. Kapansin-pansin na ang mga naturang pathologies ay pinaka-karaniwan sa mga bata at kabataan.
Neurofibroma (nagpapatong na mga sintomas: 2 sa 11)
Ang Neurofibroma ay isang benign tumor na nabubuo mula sa mga nerve tissue sa parehong central at peripheral nervous system. Sa karamihan ng mga sitwasyon, ito ay nasuri sa mga kabataan at nasa katanghaliang-gulang na mga tao.
Scheuermann-Mau disease (nagpapatong na mga sintomas: 2 sa 11)
Ang sakit na Scheuermann-Mau (syn. Scheuermann's kyphosis, dorsal (dorsal) juvenile kyphosis) ay isang progresibong deformity ng spinal column na nabubuo sa panahon ng aktibong paglaki ng katawan. Kung walang napapanahong paggamot, maaari itong humantong sa malubhang kahihinatnan.
Melanosis (nagkakasabay na sintomas: 2 sa 11)
Ang melanosis ay isang malaking halaga ng melanin pigment na naipon sa katawan. Lumalabas ang malakas na pigmentation sa ilang bahagi ng balat. Kadalasan, ang patolohiya ay nasuri sa mga kababaihan ng edad ng panganganak. Ang problema ay nagiging sanhi ng kakulangan sa ginhawa, dahil ito ay nakikita sa paningin, ang paggamot ay mahirap. Anumang therapy na sinimulan ay magdadala lamang ng kaunting ginhawa, at ang mga pagbabago sa pamumuhay ay madalas na pinapayuhan bilang isang hakbang sa pag-iwas.
Pinagmulan
Mga diagnostic

Ang Albright's syndrome ay nakikilala hindi lamang sa pamamagitan ng paglaban ng mga bato at balangkas sa parathyroid hormone, kundi pati na rin ng iba pang mga organo sa kaukulang mga hormone - ang mga ovary sa gonadotropic, ang thyroid gland sa thyrotropic, ang adrenal glands sa antidiuretic. Ang Albright's syndrome ay sinamahan ng mga sumusunod na endocrinopathies: hyperthyroidism, acromegaly, Itsenko-Cushing's syndrome.
- Ang thyroid dysfunction ay ipinakikita ng tinatawag na goiter, nodules at cysts ng organ.
- Ang labis na produksyon ng growth hormone ng pituitary gland ay humahantong sa pag-unlad ng acromegaly. Ang mga tampok ng mukha ay nagiging magaspang, ang mga braso at binti ay nagiging mahaba, ang arthritis ay nangyayari.
- Ang labis na pagtatago ng mga adrenal hormone ay nagdudulot ng labis na katabaan, pagkabansot sa paglaki, at pagkasira ng balat - mga tipikal na katangian ng Cushing's syndrome.
Mayroon ding mas mataas na dalas ng mga sakit na autoimmune, diabetes mellitus, mga depekto sa vascular, osteodysplasia ng cartilage.
Kabilang sa mga komplikasyon ng Albright's syndrome ang: optic nerve atrophy, exophthalmos, pathological fractures, bone deformity, malignancy of fibrous foci, hemorrhages sa iba't ibang bahagi ng katawan, pagdurugo, pagkagambala sa bituka, atay, bato, at digestive system.
Ang pagbabala ng therapy na may napapanahong pagkilala sa Albright's disease ay medyo kanais-nais, ngunit imposibleng ganap na pagalingin ang isang namamana na depekto ng enzyme. Ang normalisasyon ng nilalaman ng calcium at phosphorus sa katawan ay magpapahintulot sa mga pasyente na makisali sa magaan na trabaho, hindi kasama ang nervous strain at magtrabaho kasama ang mga makina at mekanismo.

Sa paulit-ulit na pag-atake ng muscle cramps na mahirap gamutin sa pamamagitan ng gamot, ang medikal na komisyon ay nagpasya sa pagtatalaga ng isang grupong may kapansanan. Sa mga malubhang kaso ng pseudohypoparathyroidism, maaaring magkaroon ng mga sumusunod na komplikasyon:

Ang pagpapalitan ng calcium at phosphorus sa katawan
Ang metabolismo ay ang pinakamahalagang aspeto ng buhay ng katawan ng tao. Bawat segundo sa mga tisyu at organo, ang ilang mga kemikal na elemento ay na-convert sa iba. Bilang isang patakaran, ang kanilang nilalaman ay mahigpit na kinokontrol. Ito ay totoo lalo na sa mga electrolyte - sodium, potassium, calcium at phosphorus. Ito ay salamat sa kanilang mga tiyak na ratios na ang gawain ng puso at mga kalamnan ng kalansay, ang paghahatid ng isang nervous electrical signal, ay posible.
Ang kaltsyum ay matatagpuan sa lahat ng mga organo at tisyu at gumaganap ng ilang mahahalagang tungkulin:

Ang posporus ay matatagpuan sa malalaking dami sa tissue ng buto at mahalaga para sa normal na paggana ng utak. Sa isang mahigpit na tinukoy na ratio, ang calcium at phosphorus ay naroroon sa dugo. Ang labis ng mga kemikal na compound na ito ay pinalalabas ng mga bato sa ihi.
Ang parathyroid hormone ay ang pangunahing regulator ng mga antas ng calcium at phosphorus sa dugo.

Ang kagalingan ng katawan ay direktang nakasalalay sa isang pare-parehong halaga ng calcium at phosphorus. Ang proseso ng kanilang metabolismo ay kinokontrol ng thyroid at parathyroid glands:
- Ang una ay gumagawa ng hormone calcitonin, dahil sa kung saan ang calcium mula sa dugo ay pumasa sa komposisyon ng tissue ng buto.
- Ang huli, sa tulong ng parathyroid hormone, ay nag-aalis ng kakulangan ng calcium sa dugo sa pamamagitan ng paghuhugas nito sa mga buto.
Bilang karagdagan, ang pagpapalitan ng calcium at phosphorus ay kinokontrol ng mga bato, na nag-aalis ng labis sa mga kemikal na compound na ito sa ihi. Malaki ang papel na ginagampanan ng bitamina D dito.
Ang Albright's syndrome ay isang namamana na sakit na sanhi ng isang depekto sa metabolismo ng calcium at phosphorus sa katawan. Ito ay humahantong sa iba't ibang pagbabago sa mga buto, bato, digestive tract at mental disorder.
Ang namamana na osteodystrophy ay unang isinasaalang-alang ng siyentipikong si Fuller Albright noong 1942. Ang sakit ay ilang beses na mas karaniwan sa mga kababaihan. Mga 300 kaso lamang ng bihirang patolohiya na ito ang inilarawan sa medikal na panitikan.
Pag-uuri
Ang pseudohypoparathyroidism ay nahahati sa tatlong pangunahing uri:

Mga pamamaraan ng diagnostic
Upang makilala ang Albright's disease, ang magkasanib na gawain ng ilang mga espesyalista ay kinakailangan: isang urologist, isang neurologist, isang psychiatrist, isang endocrinologist, at isang geneticist. Upang maitatag ang tamang diagnosis, ginagamit ang mga sumusunod na pamamaraan:

Ang pagkakaiba-iba ng diagnosis ay isinasagawa sa mga sumusunod na sakit:

Ang pseudohypoparathyroidism ay nangangailangan ng panghabambuhay na follow-up ng isang endocrinologist. Sa kaso ng paulit-ulit na malubhang seizure ng mga kombulsyon, ang pagpapaospital sa espesyal na departamento ng ospital ay kinakailangan.
Ang paggamot sa mga gamot ay ang pangunahing bahagi ng paggamot ng pseudohypoparathyroidism. Ang mga pangunahing gawain ay:
- normalisasyon ng antas ng kaltsyum at posporus sa dugo;
- pagpapanumbalik ng normal na istraktura ng tissue ng buto;
- pag-iwas sa mga seizure;
- pinipigilan ang pagbuo ng mga bato sa bato.
Ang calcium chloride at calcium gluconate ay ginagamit upang gawing normal ang mga antas ng calcium. Kadalasan, ang mga gamot na ito ay ginagamit sa anyo ng mga tablet. Gayunpaman, upang maalis ang mga cramp ng kalamnan, ang intravenous administration ng mga pharmacological agent ay isinasagawa.
Ang Calcium gluconate ay isang gamot na ginagamit upang gamutin ang Albright's disease.
Ang mga paghahanda ng bitamina D ay nag-aambag sa normalisasyon ng antas ng calcium at phosphorus sa dugo:
- Kaltsyum D3;
- Oksidevit;
- Calcitrin.
Ang Calcium D3 ay inireseta para sa Albright's disease
Ang appointment ng parathyroid hormone sa pseudohypoparathyroidism ay hindi epektibo, dahil ang mga tisyu at organo ay hindi sensitibo sa impluwensya nito.
Pagwawasto ng diyeta
Ang isang mahalagang bahagi ng paggamot ng pseudohypoparathyroidism ay ang pagbabago sa pandiyeta. Kasabay nito, ang sapat na calorie at fractional na nutrisyon ay mahalaga. Ang dami ng likido na lasing bawat araw ay dapat na hindi bababa sa 1.5-2 litro.
- beans;
- berdeng gisantes;
- lentil;
- pili;
- watercress;
- kintsay;
- kuliplor;
- dill;
- basil;
- gatas;
- yogurt;
- kulay-gatas.
Ang Albright's syndrome ay isang bihirang sakit na minana at ipinakikita ng pinsala sa skeletal system. Ang dahilan para dito ay isang paglabag sa metabolismo ng calcium at phosphorus. Ito ay nangyayari dahil sa paglaban ng mga tisyu sa parathyroid hormone, na ginawa ng mga glandula ng parathyroid. Ang sindrom na ito ay kadalasang sinasamahan ng mental at pisikal na pag-unlad ng retardation.
Ang sintomas na ito ay tinatawag ding pseudohypoparathyroidism.
Sa kauna-unahang pagkakataon ang sakit na ito ay inilarawan ng siyentipikong si Albright, kung saan nakuha nito ang pangalan nito.
Mga uri
Mayroong dalawang uri ng pseudohypoparathyroidism, depende sa antas ng calcium sa dugo:
- Ang unang klinikal na larawan ay halos kapareho sa idiopathic hypoparathyroidism - mababang antas ng kaltsyum at insensitivity sa parathyroid hormone.
- Ang pangalawa ay nagpapakita ng isang normal na antas ng calcium, samakatuwid ito ay tinatawag na pseudohypoparathyroidism.
Sinasabi ng mga doktor na ang isang anyo ng sakit ay maaaring mapunta sa isa pa. Minsan sa mga miyembro ng pamilya ang sakit na ito ay maaaring magkaroon ng iba't ibang anyo.
Sa mga lalaki, ang patolohiya na ito ay nagdudulot ng reproductive dysfunction, kaya bihira itong maipadala sa pamamagitan ng male line.
Mga sanhi at pag-unlad
Ang genetic na patolohiya na ito ay nailalarawan sa pamamagitan ng isang autosomal na nangingibabaw na mode ng mana.
Ang pangunahing dahilan ay isang depekto ng kapanganakan na ginagawang hindi sensitibo ang mga peripheral tissue sa parathyroid hormone.
Depende sa kung paano at kung saan nasira ang metabolic chain, dalawang uri ng sindrom ay nakikilala.
palatandaan
Ang mga karaniwang nakikitang palatandaan ay kinabibilangan ng:
- mababang paglago;
- sobra sa timbang;
- mataas na antas ng glucose;
- mukha ng buwan;
- mga karamdaman sa pagdadalaga.
Ang mga sumusunod na sintomas ay maaari ding lumitaw:
- acromegaly;
- mga sakit sa autoimmune;
- gynecomastia;
- Itsenko-Cushing's disease, atbp.
Ang mga pasyente ay madalas na nagreklamo ng dugo sa ihi, patuloy na pagsusuka, pagbabago sa ngipin, kombulsyon. Sa pagsusuri, makikita ang subcutaneous, at maaaring lumitaw ang mga calcification.
Kadalasan, ang mga pasyente ay pinaikli ang mga daliri, ang mga epiphyses ng tubular bones ay deformed, at maaaring may mga palatandaan ng isang lenticular cataract.
Halos palaging, ang mga naturang pasyente ay nahuhuli sa pag-unlad ng kaisipan.
Mga diagnostic
Ang sakit ay tinukoy sa mga batang may edad na lima hanggang sampung taon. Ang doktor ay tumatanggap ng isang katangian ng klinikal na larawan, nagpapakita ng isang malaking bilang ng mga anomalya sa pag-unlad ng balangkas, ang pagkakaroon ng:
- hypocalcemia;
- hyperphosphatemia;
- nabawasan ang paglabas ng calcium at phosphorus sa ihi;
- nadagdagan ang nilalaman ng parathyroid hormone;
- renal tubular resistance, atbp.
Paggamot
Ang paggamot ay binubuo ng iba't ibang paghahanda ng calcium, na partikular na pinili upang mapanatili ang homeostasis sa isang normal na estado.
Kinakailangan din na ipakilala ang mga bitamina ng pangkat D, calcitriol, oxydevit.
Ang mga indibidwal ay bumuo ng diyeta na naglilimita sa mga pagkaing mataas sa posporus at nagtataguyod ng normal na antas ng calcium sa dugo.
Kung may mga magkakatulad na sakit ng endocrine system, ang isang bilang ng mga hormonal na gamot ay inireseta din bilang kapalit na therapy.
Ang mga seizure ay inaalis gamit ang calcium gluconate o calcium chloride solution.
Kung lapitan mo ang paggamot nang komprehensibo at tama, maaari itong magbigay ng isang kanais-nais na resulta. Ngunit ang bawat ganoong tao, kahit na siya ay gumaling at namumuhay ng normal, bago magkaroon ng anak, ay dapat suriin ng isang geneticist upang matukoy ang mga panganib.
Sa unang pagkakataon sa panitikang Ruso, ang sindrom na ito ay inilarawan ni V. R. Braytsev noong 1922, na tinukoy ito bilang "fibrous tumors". Pagkalipas ng labinlimang taon, binuo ng endocrinologist na si Fuller Albright ang mga palatandaan ng sistematikong sakit na ito tulad ng sumusunod: "isang sindrom na nailalarawan sa pamamagitan ng disseminated osteitis fibrous, pigmentation fields at endocrine disorder na may maagang pagbibinata sa mga batang babae."
Sa madaling salita, ang McCune-Albright-Braytsev syndrome ay isang sistematikong sakit na nailalarawan sa napaaga na sekswal na pag-unlad at patolohiya ng buto. Ang sakit na ito ay nangyayari nakararami sa mga babae at isang congenital hereditary pathology. Hanggang kamakailan lamang, pinaniniwalaan na ito ay isang eksklusibong sakit na babae, ngunit pagkatapos ay naitala din ang mga kaso ng mga sakit sa mga lalaki. Ang sanhi ng pag-unlad ng sindrom ay itinuturing na isang mutation sa isang partikular na gene.
Mga palatandaan ng McCune-Albright-Braytsev syndrome
- Mga dark spot. Ang lahat ng mga pasyente ay may espesyal na pigmentation ng isang hindi regular na hugis, ang kulay ng kape na may gatas. Ang mga batik ay karaniwang matatagpuan sa likod, dibdib, hita, pigi at ibabang likod. Ito ay isang uri ng marker ng sakit, ang pigmentation ay naroroon mula sa kapanganakan o lumilitaw habang lumalaki ang bata.
- Patolohiya ng buto. Ang mahahabang tubular bones (hal., femurs) ay kadalasang apektado. Ang pagkakaroon ng mga cavity sa mga buto ay humahantong sa kanilang pagpapapangit, ang paglitaw ng mga bali at kurbada ng mga limbs. Karaniwan ang baluktot na pagpapapangit ng pastol. Posibleng pinsala sa mga buto ng bungo. Nagdudulot ito ng asymmetry ng mukha, unilateral exophthalmos (bulging eyes). Ang pagkasira ng buto at pagkabali ay bumababa pagkatapos ng pagdadalaga.
- Precocious sekswal na pag-unlad. Maaari itong magsimula sa edad na 6-9 na buwan at hanggang 7 taon. Kadalasan, ang mga unang palatandaan ng pagdadalaga ay lumilitaw sa isang bata sa 3 taong gulang. Maaari itong lumitaw sa buo at hindi kumpletong mga anyo. Ang buong anyo sa mga batang babae ay nailalarawan sa pamamagitan ng menarche (unang regla) at pagpapalaki ng dibdib. Posible ang matagal at labis na pagdurugo, madalas na matatagpuan ang mga cyst sa mga ovary. Sa hindi kumpletong anyo, hindi nangyayari ang menarche.
Ang napaaga na sekswal na pag-unlad sa mga lalaki ay hindi gaanong karaniwan, na sinamahan ng isang simetriko na pagtaas sa mga testicle, pagkatapos ay ang paglaki ng ari ng lalaki at pubic hair, gayundin sa normal na sekswal na pag-unlad.
- Pagpapabilis ng edad ng buto at pinabilis na pagsasara ng mga zone ng paglago.
- Minsan may mga neurological at mental disorder (mental retardation, convulsions, atrophy ng optic nerve, pagkawala ng pandinig).
- Ang panitikan ay naglalaman ng impormasyon tungkol sa iba pang mga endocrine disorder sa sakit na ito: acromegaly, hyperparathyroidism (nadagdagang pag-andar ng mga glandula ng parathyroid), hyperthyroidism (nadagdagang pag-andar ng thyroid gland) at ang pagkakaroon ng mga node sa thyroid gland, hypercorticism (isang labis na mga glucocorticoid hormones ng adrenal glands), rickets na lumalaban sa bitamina D.
Diagnosis ng McCune-Albright-Braytsev syndrome
- Ang pagkakaroon ng mga tampok na katangian.
- konsultasyon sa genetika.
- X-ray na pagsusuri.
- Mga sex hormone at gonadotropin, thyroid hormone, prolactin, iba pang pituitary tropic hormones (ayon sa mga indikasyon).
- Ultrasound ng pelvic organs para sa mga babae at scrotum para sa mga lalaki.
- Konsultasyon ng isang endocrinologist at isang pediatric gynecologist para sa mga batang babae o isang pediatric urologist para sa mga lalaki.
- Pagsusuri ng iba pang mga organo ng endocrine system (ayon sa mga indikasyon).
Paggamot ng McCune-Albright-Braytsev syndrome
Ang mga pasyente na may ganitong patolohiya ay nangangailangan ng pangangasiwa ng isang gynecologist-endocrinologist, endocrinologist, radiologist, ophthalmologist, orthopedic traumatologist. Bilang karagdagan, kinakailangan na pana-panahong matukoy ang antas ng tropiko at gonadotropic hormones. Walang tiyak na paggamot para sa sindrom na ito. Kadalasan, pinipili ng mga doktor ang mga taktika sa pagmamasid. Para sa mga batang babae, ang therapy ay ipinahiwatig sa kaso ng isang matagal na pagtaas sa mga antas ng estrogen, na sinamahan ng madalas at matinding pagdurugo. Ayon sa mga indikasyon, ang pagwawasto ng mga deformidad ng buto ay isinasagawa. Sa pag-unlad ng mga sintomas ng pinsala sa iba pang mga glandula ng endocrine, ang paggamot ay isinasagawa ng mga endocrinologist.
Catad_tema Pediatrics - Mga Artikulo
Pseudohypoparathyroidism (Ang namamana na osteodystrophy ni Albright): mga kahirapan sa paghahanap ng kaugalian na diagnostic. Klinikal na pagmamasid
E.V. Tozliyan, pediatric endocrinologist, geneticist, PhD, I.V. Shulyakova, neurologist, PhD,
Hiwalay na structural subdivision "Scientific Research Clinical Institute of Pediatrics" SBEE HPE "Russian National Research Medical University na pinangalanan sa N.I. Pirogov" ng Ministry of Health ng Russian Federation, Moscow
Mga keyword:
mga bata, pseudohypoparathyroidism, Albright hereditary osteodystrophy, obesity, hypocalcemia, diagnosis, paglaban sa parathyroid hormone.
mga keyword: mga bata, pseudohypoparathyroidism, Albright hereditary osteodystrophy, obesity, hypocalcemia, diagnostics, parathyroid hormone resistance.
Pseudohypoparathyroidism (Greek pseudes - false + hypoparathyroidism; kasingkahulugan: Albright's hereditary osteodystrophy, "Javanese chicken" syndrome) ay isang bihirang namamana na sakit ng skeletal system na ginagaya ang hypoparathyroidism at nailalarawan sa pamamagitan ng kapansanan sa metabolismo ng calcium at phosphorus; madalas na sinamahan ng pagkaantala sa mental at pisikal na pag-unlad. Ang sakit ay unang inilarawan ng American endocrinologist na si Albright F. noong 1942. Ang pagkalat ng sakit ay 7.9 bawat 1 milyong tao.
GENETIC DATA
Ang Pseudohypoparathyroidism (PHP) ay isang genetically heterogenous na sakit. Ang data sa uri ng namamana na paghahatid ay magkasalungat: parehong X-linked dominant at autosomal dominant, autosomal recessive na mga uri. Sa karamihan ng mga kaso, ang pagbuo ng namamana na osteodystrophy ni Albright ay nauugnay sa mga mutasyon sa locus 20q13 ng GNAS1 gene na matatagpuan sa chromosome 20 (Patten et al., 1990), na nag-encode ng Gs-alpha protein na nauugnay sa parathyroid hormone (PTH) receptor. Ang isang katulad na phenotype ay natagpuan din sa mga pasyente na may interstitial na pagtanggal ng mahabang braso ng chromosome 2 ng 2q37 locus.
PATHOGENESIS
Ang pathogenesis ng pseudohypoparathyroidism ay batay sa genetically natukoy na paglaban ng mga bato at balangkas sa pagkilos ng parathormone bilang isang resulta ng isang depekto sa kumplikadong "specific cytoreceptor - parathyroid hormone - adenylate cyclase", na nakakagambala sa pagbuo ng cyclic 3 "- , 5"-adenosine monophosphate (cAMP) sa mga bato, na siyang intracellular na tagapamagitan ng pagkilos ng parathyroid hormone sa mga metabolic na proseso. Ang pseudohypoparathyroidism ay genetically magkakaibang sakit. Sa ilang mga pasyente, ang cytoreceptor mismo ay may depekto, na nagbubuklod sa parathyroid hormone (type 1A pseudohypoparathyroidism), habang ang iba ay may depekto sa nucleotide-binding protein na naisalokal sa lipid bilayer ng cell membrane at gumaganang nagbubuklod sa receptor sa adenylate cyclase (uri. 1B pseudohypoparathyroidism). Ang ilang mga pasyente ay may enzymatic deficiency ng adenylate cyclase mismo (pseudohypoparathyroidism type 2). Ang kakulangan ng cAMP, na nagreresulta mula sa mga depektong ito, ay humahantong sa isang paglabag sa synthesis ng mga partikular na protina na tumutukoy sa biological na epekto ng parathyroid hormone. Kaya, ang sensitivity ng mga target na organo sa parathyroid hormone ay nawala.
MGA KLINIKAL NA KATANGIAN
Sa kasalukuyan, 4 na klinikal na anyo ng patolohiya ang nakikilala: mga uri 1A, 1B, 1C at 2. Ang kaalaman sa kanilang mga klinikal at biochemical na tampok at data ng genetic na pananaliksik ay nagpapahintulot sa differential diagnosis sa loob ng nosological form mismo.
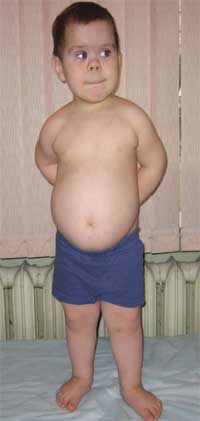
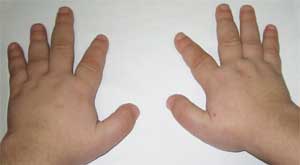
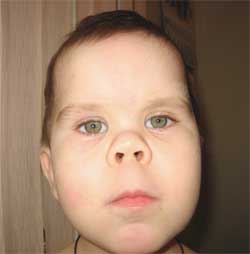
Ang mga pangkalahatang palatandaan na nagpapahintulot sa paghihinala sa sakit ay hindi katimbang ng pisikal na pag-unlad, maikling tangkad (hanggang sa dwarfism) dahil sa pagpapaikli ng mas mababang paa (larawan 1), brachydactyly (larawan 2), bilog na "hugis-buwan" na mukha (larawan 3). Minsan may mga exostoses at aplasia ng ngipin.
Larawan 1.
Hitsura ng isang bata na may Albright osteodystrophy
(mga tampok ng phenotype, maikling tangkad dahil sa pag-ikli ng lower limbs)
Larawan 2.
Mga tampok ng skeletal system sa isang pasyente
na may Albright osteodystrophy
(brachydactyly - maiikling daliri)
Larawan 3.
Mga tampok ng phenotype ng bata
na may Albright osteodystrophy
(bilog na mukha ng "buwan")
Ang isang matalim na pagpapaikli ng I, III at V metacarpal at metatarsal bones (lalo na III at IV) ay itinuturing na isang pathognomonic sign, bilang isang resulta kung saan ang mga daliri ng II sa mga kamay at paa ay mas mahaba kaysa sa iba, at kapag ang kamay ay nakakuyom sa isang kamao, walang mga bulge sa rehiyon ng IV at V metacarpophalangeal joints - ang tinatawag na brachymetaphalangism. Ang mga maikling malawak na phalanges, pampalapot ng cranial vault at demineralization ng mga buto (osteoporosis), ang labis na katabaan ay napansin din.
Ang mental retardation (kadalasan sa katamtamang kalubhaan) ay matatagpuan sa humigit-kumulang 20% ng mga pasyente. Ayon sa ilang mga may-akda, ang oligophrenia ay nangyayari sa 70% ng mga kaso na may hypocalcemia at sa 30% ng mga kaso na may normocalcemia. Ang mga proseso ng pag-iisip sa mga pasyente ay pinabagal. Sa neurological status, motor awkwardness, neurotic reaksyon ay madalas na nabanggit: takot, pagkabalisa, pagkabalisa, mahinang pagtulog, nadagdagan reflexes, convulsions na tetanic at sanhi ng hypocalcemia, minsan convulsive paroxysms. Ang mga sintomas ng myopathic ay inilarawan din: pagkapagod ng kalamnan, kahinaan ng kalamnan. Ang mga extrapyramidal disorder ay madalas na sinusunod: choreiform hyperkinesis, athetosis, facial hemispasm, parkinsonism, sa ilang mga kaso mayroong epileptic paroxysms, cerebellar sintomas: ataxia, may kapansanan sa koordinasyon.
Kadalasan, ang mga soft tissue calcifications, subcutaneous calcifications (dibdib, tiyan, calcaneal tendons) ay tinutukoy, na may histological na pagsusuri kung saan - osteoma cutis(Izraeli et al., 1992), utak (basal ganglia). Mahalagang tandaan na ang mga calcification ay maaaring naroroon na sa kapanganakan. Dahil sa hypocalcemia, kadalasang nagkakaroon ng mga katarata at nangyayari ang mga depekto sa enamel ng ngipin.
PSEUDOHYPOPARATHYROISIS TYPE 1A
ay may autosomal dominant mode ng inheritance. Ang type 1A pseudohypoparathyroidism gene, GNAS1, ay matatagpuan sa mahabang braso ng chromosome 20, sa 20q13.2 locus. Ang pag-unlad ng sakit ay nauugnay sa isang kakulangan ng guanine-nucleotide-binding protein (Gs-protein). Kasabay nito, ang PTH, na nagbubuklod sa mga receptor ng mga target na tisyu, ay hindi nakakapag-activate ng cyclic adenosine monophosphate (cAMP) at nagiging sanhi ng pagtugon sa tissue. Marahil, ang isang katulad na mekanismo ay sumasailalim sa pagbuo ng insensitivity ng mga tisyu ng iba pang mga organo at mga glandula ng endocrine (hypofunction ng thyroid gland, gonads, pituitary gland, diabetes mellitus, pati na rin ang isang pinababang tugon ng atay sa pangangasiwa ng glucagon) na sinusunod sa type 1A pseudohypoparathyroidism. Sa ganitong uri ng patolohiya, walang tumaas na paglabas ng cAMP sa ihi, na katangian ng pamantayan, bilang tugon sa exogenous administration ng PTH. Ang sakit ay mas madalas na nasuri sa edad na 5-10 taon. Ang mga pasyente ay may maikling tangkad, isang maikling leeg, isang bilog na mukha, pagpapaikli ng metacarpal at metatarsal na mga buto (mas madalas na pagpapaikli ng IV at mas madalas na mga daliri ng II) - ang tinatawag na brachy-metaphalangism. Ang pag-calcification ng malambot na mga tisyu, ang mga subcutaneous calcifications ay nabanggit, na maaaring makita na sa kapanganakan; madalas na may sabay-sabay na paglahok ng iba pang mga glandula ng endocrine: ang thyroid gland (hypofunction), gonads, pancreas (diabetes mellitus). Dahil sa hypocalcemia, madalas na nagkakaroon ng mga katarata at depekto sa enamel ng ngipin. Bilang isang differential diagnostic test para sa pagkakaiba sa pagitan ng PPH type 1A at hypoparathyroidism: ang kawalan ng isang klinikal na epekto mula sa parenteral administration ng PTH sa anyo ng isang pagtaas sa antas ng calcium sa dugo at isang pagtaas sa renal excretion ng phosphorus sa ihi (phosphaturic effect).
Ang biochemical examination ay nagpapakita ng hypocalcemia, hyperphosphatemia, isang pagtaas sa antas ng parathyroid hormone sa dugo, at hypophosphaturia. Ang antas ng Gs-protein sa dugo ay nabawasan. Ang pagsusuri sa X-ray ng skeletal system ay nagpapakita ng pag-ikli ng metacarpal at metatarsal bones, pangkalahatang demineralization, at pagpapalapot ng mga buto ng cranial vault.
PSEUDOHYPOPARATHYROISIS TYPE 1B
ay may autosomal dominant na uri ng mana, gayunpaman, ang isang nangingibabaw, X-linked na uri ng mana ay hindi ibinubukod. Kinakailangang tandaan ang minsang sinusunod na hindi kumpletong pagtagos ng gene ng sakit at ang posibilidad ng isang nakatagong karwahe ng patolohiya. Samakatuwid, inirerekumenda ang clinical (detection ng subclinical course ng sakit) at biochemical examination (pagtukoy ng mga antas ng calcium, phosphorus, blood PTH) ng mga di-umano'y carrier ng sakit. Ang HPT type 1B ay sanhi ng kakulangan ng tissue receptors para sa parathyroid hormone sa mga target na organo at limitadong resistensya sa parathyroid hormone. Ang klinikal na larawan ay katulad ng uri ng 1A na klinika, ngunit walang pinsala sa iba pang mga glandula ng endocrine, at ang osteodystrophy ay hindi gaanong karaniwan.
Sa mga pasyente, walang reaksyon sa bato sa exogenous na pangangasiwa ng parathyroid hormone sa anyo ng isang pagtaas sa excretion ng cyclic adenosine monophosphate sa ihi, gayunpaman, hindi katulad ng type 1A, ang antas ng Gs-protein sa dugo ay normal. Ang mga kababaihan ay mas madalas na apektado kaysa sa mga lalaki, ngunit ang kalubhaan ng sakit ay maaaring pareho sa mga kalalakihan at kababaihan.
PSEUDOHYPOPARATHYROISIS TYPE 1C
ang ilang mga may-akda ay tumutukoy sa pseudo-pseudohypoparathyroidism (PPHP) na inilarawan ni Albright F. noong 1952. Ito ay nailalarawan sa pamamagitan ng isang klinikal na larawan na katangian ng PAH, ngunit ang mga antas ng calcium, phosphorus sa dugo at ihi ay nananatili sa loob ng normal na hanay. Ang mga tagapagpahiwatig ng PTH at Gs-protein sa dugo ay nananatili rin sa isang normal na antas. Ang ilang mga pasyente na may PHP type 1C ay may mga pagtanggal de novo sa chromosome 2. Posible na ang variant na ito ng sakit ay isang subtype ng HPP type 1A.
PSEUDOHYPOPARATHYROISIS TYPE 2
klinikal na katulad sa iba pang mga uri ng sakit, ngunit may isang autosomal recessive mode ng mana. Ang pagkakaroon ng mga autosomal na nangingibabaw na anyo ng patolohiya ay hindi ibinukod. Ang pathogenesis ng pag-unlad ay nauugnay sa intracellular resistance sa cAMP. Sa kasong ito, ang PTH ay nagbubuklod sa mga receptor at nagiging sanhi ng isang normal na tugon ng cell sa PTH sa anyo ng pagtaas sa paglabas ng cAMP. Ang intracellular insensitivity sa cAMP, gayunpaman, ay hindi pinapayagan ang buong pagsasakatuparan ng pagkilos ng PTH. Kasabay nito, ang normal na reaksyon ng mga bato sa exogenous na pangangasiwa ng parathyroid hormone ay napanatili sa anyo ng isang pagtaas sa paglabas ng cyclic adenosine monophosphate sa ihi. Iminungkahi na ang uri 2 HWP ay maaaring nauugnay sa kakulangan sa bitamina D.
Kaya, ang mga natukoy na uri ng PHP ay klinikal na nailalarawan sa pamamagitan ng nabawasan na sensitivity ng mga target na organo sa PTH, ngunit naiiba sa mga pathogenetic na mekanismo ng pagbuo ng tissue insensitivity.
DIAGNOSTICS
Ang likas na katangian ng renal cAMP excretion bilang tugon sa pangangasiwa ng PTH ay maaaring magsilbi bilang isang laboratory differential diagnostic test: ang pagtaas ng cAMP excretion ay nabanggit sa type 2 at ang kawalan nito sa type 1. Ang diagnosis ay nakumpirma sa pamamagitan ng pagtuklas ng isang pinababang antas ng guanine-nucleotide -nagbubuklod na protina (Gs-protein) sa dugo (sa average na 1.5-2 beses) kumpara sa pamantayan. Ang hypocalcemia ay kadalasang nauugnay sa hyperphosphatemia at hypophosphaturia. Ang antas ng PTH ay nakataas; na may uri 1C, ang antas ng PTH ay normal, na nagbunga ng pangalang "pseudohypoparathyroidism". Ang pagsusuri sa x-ray ng skeletal system ay nagpapakita ng pag-ikli ng metacarpal at metatarsal bones, kadalasang pangkalahatan demineralization (osteoporosis), pampalapot ng mga buto ng cranial vault. Ang dermatoglyphic drawing ay nagpapakita ng displacement ng axial palmar triradius.
Pamantayan sa Diagnosis:
- mababang paglago;
- bilugang mukha;
- naantala ang pag-unlad ng neuropsychic;
- mga anomalya ng kalansay;
- mababang serum calcium;
- mataas na antas ng parathyroid hormone sa dugo;
- nabawasan ang paglabas ng phosphate at cAMP sa ihi.
PAGGAgamot at pag-iwas
Ang paggamot para sa hypocalcemia ay binubuo sa pagtatalaga ng mga suplementong calcium sa mga dosis na sapat upang mapanatili ang isang normal na konsentrasyon ng calcium sa dugo. Napakahalaga ng therapy sa bitamina D. Sa kasalukuyan, ginagamit ang mga aktibong metabolite ng bitamina D - oxidevit, 1-alpha-D3, calcitrin, atbp. sa isang dosis na 1-2 mcg / araw na may positibong resulta (nadagdagan ang calcium sa dugo , nabawasan ang mga pagpapakita ng convulsive syndrome ). Ang tachystin (0.5–1.5 mg/araw) ay epektibo rin. Pinapataas ng gamot na ito ang pagsipsip ng calcium sa bituka at sa gayon ay pinapataas ang antas ng calcium sa dugo. Ang anticonvulsant therapy ay ginagamit bilang pandagdag na paggamot. Ang paggamot ay walang kapansin-pansing epekto sa pag-unlad ng intelektwal, ngunit kasama ang pagbawas sa mga sintomas ng convulsive syndrome, ang isang regression ng neurological manifestations (subcortical disorders, choreiform hyperkinesis, athetosis, atbp.) Ay sinusunod. Upang maiwasan ang labis na dosis ng mga paghahanda ng bitamina D, kinakailangan na kontrolin ang konsentrasyon ng calcium sa dugo tuwing 3-7 araw sa unang 2 linggo ng paggamot at bawat buwan para sa susunod na 2-3 buwan. Sa pag-abot sa isang matatag na konsentrasyon ng calcium sa dugo, sapat na upang suriin ito isang beses bawat 2-3 buwan. Ang phosphorus-restricted diet ay nakakatulong na gawing normal ang parehong konsentrasyon ng phosphorus at calcium sa dugo at alisin ang mga sintomas ng pangalawang hyperparathyroidism. Sa kaso ng kakulangan ng iba pang mga glandula ng endocrine, isinasagawa ang kapalit na therapy na may naaangkop na mga hormone.
Ang paggamot na may parathyroid hormone ay hindi epektibo. Para sa kaluwagan ng convulsive na pag-atake, ang isang 10% na solusyon ng calcium chloride o calcium gluconate ay ibinibigay sa intravenously; sa loob - 5-10% na solusyon ng calcium chloride, 1 kutsara 3-4 beses sa isang araw: calcium gluconate, calcium lactate - hanggang 10 g bawat araw.
PAGTATAYA para sa buhay ay tinutukoy ng kalubhaan ng convulsive syndrome.
PAG-Iwas ang sakit ay batay sa data ng medikal na genetic counseling.
MEDICAL GENETIC COUNSELING
Sa medikal na genetic counseling, ang isa ay dapat magpatuloy mula sa autosomal dominant na uri ng mana at isang mataas na (50%) na panganib ng pag-ulit ng sakit sa pamilya na may minanang anyo. Upang matukoy ang likas na katangian ng uri ng mana, kinakailangan na magsagawa ng masusing pagsusuri sa mga magulang, dahil ang sindrom ay maaaring magpakita mismo na may kaunting mga klinikal na sintomas. Sa kasalukuyan, ang molecular genetic diagnosis ng sakit ay binuo at pinapabuti sa pamamagitan ng pag-type ng mga mutasyon sa GNAS1 gene sa chromosome 20. Ang mga pamamaraan para sa prenatal diagnosis ng sakit sa kabuuan at ang mga indibidwal na uri nito ay binuo.
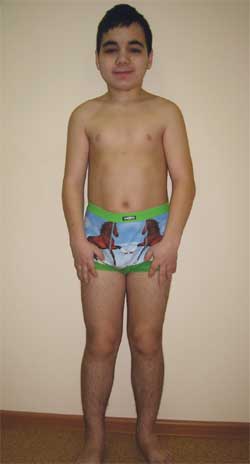
CLINICAL OBSERVATION Si Boy G., 14.5 taong gulang (larawan 4), ay ipinasok sa Research Clinical Institute of Pediatrics na may diagnosis ng isang degenerative disease ng nervous system? congenital panlabas na hydrocephalus; nagpapakilala ng epilepsy; hereditary syndrome? sakit sa imbakan? metabolic encephalopathy; subclinical hypothyroidism; maikling tangkad ng mixed genesis; kapansanan sa pag-iisip.
Mga reklamo sa pagpasok sa matinding paroxysmal sakit ng ulo, naisalokal sa frontal na rehiyon at sinamahan ng pagsusuka, na nagdudulot ng kaluwagan, nabawasan ang memorya at pagganap ng paaralan, mga convulsive na pag-atake, kung saan ang pagkibot ay nangyayari sa kanang kamay.
Larawan 4.
Batang G., 14.5 taong gulang, na may Albright osteodystrophy
(mga tampok ng phenotype, maikling tangkad, pagpapaikli ng mga limbs, brachydactyly)
Kasaysayan ng pamilya: ang mga magulang ay mga Armenian ayon sa nasyonalidad, hindi nauugnay sa dugo at walang mga panganib na propesyonal. Walang mga kaso ng sakit sa isip, epilepsy, pagkaantala sa pag-unlad sa pedigree. Mga kapatid, kapatid na babae, 17 taong gulang - mula sa mga salitang - malusog.
Kasaysayan ng buhay at sakit: isang batang lalaki mula sa ika-2 pagbubuntis, na nagpatuloy nang walang mga tampok, pangalawang kapanganakan, sa oras, physiological, timbang ng kapanganakan - 3100 g, haba - 51 cm Siya ay sumigaw kaagad, Apgar score - 7/9 puntos. Pagkasira sa ika-3 araw - neonatal convulsions, tumigil sa maternity hospital. Maagang postnatal period - walang mga tampok. Nagkaroon ng bahagyang pagkaantala ng tempo sa pag-unlad ng motor sa unang taon ng buhay, independiyenteng paglalakad mula sa 1 taon 3 buwan. Sa koneksyon na ito, siya ay sinusunod ng isang neurologist na may diagnosis ng organic na pinsala sa central nervous system; congenital hydrocephalus; neonatal convulsions; febrile convulsions sa kasaysayan.
Nakatanggap ng diakarb, finlepsin. Ang debut ng mga seizure mula 1 taon 11 buwan. - asymmetric, tonic sa anyo ng pag-igting ng kanang braso at binti, na may pagtatatag ng mga mata, hanggang sa 2 minuto, nang walang pagkawala ng kamalayan, madalas hanggang sa 10 na yugto bawat araw. Nakatanggap ng depakine irregularly. Laban sa background ng self-cancellation - isang solong tonic status. Sa edad na 2, ang isang CT scan ng utak ay isinagawa sa lugar ng paninirahan, kung saan ang solong foci ng demyelination ay nakita sa occipital lobes.
Kinunsulta ng isang neurosurgeon, inirerekomenda ang konserbatibong paggamot. Mula sa edad na 3 taon, mayroong pagkaantala sa pag-unlad ng psychoverbal, inirerekomenda ang pangangasiwa ng isang psychiatrist.
Mula sa edad na 4-5 taon, ang mga magulang ay nagsimulang mapansin ang pagpapapangit at pag-ikli ng mga daliri sa paa at kamay, lalo na ang mga daliri ng II-IV na simetriko sa mga kamay at paa, at pagbaba sa mga rate ng paglaki. Sa edad na 8, ang pagtatapos ng isang speech therapist ay isang pangkalahatang karamdaman sa pagsasalita ng ika-2 o ika-3 na antas, inirerekomenda ang pagsasanay sa isang dalubhasang paaralan. Sa parehong edad, pagsusuri ng isang geneticist sa lugar ng paninirahan, konklusyon: namamana na metabolic disease? ang isang pag-aaral ng mga amino acid sa dugo ay inirerekomenda, walang mga pagbabago na nakita; pangwakas na konklusyon: ang data para sa isang namamana na metabolic disease ay hindi ipinahayag; hypochondroplasia; inirerekomendang paggamot ng isang neurologist at endocrinologist.
mesa.
Profile ng pag-unlad ng kaisipan ng bata G., 14.5 taong gulang (IQ = 68)

Sa edad na 8 taon, siya ay kinonsulta ng isang endocrinologist tungkol sa paglago at pagkaantala sa pag-unlad. Kapag ang pagsusuri sa X-ray ng mga kamay, ang mga sumusunod na tampok ay nabanggit: ang gitna, pangunahing phalanges at metacarpal bones ay pinaikli at pinalapot; Ang diagnosis ng radiologist ay achondroplasia.
Paulit-ulit na sinusuri sa lugar ng paninirahan sa isang neurological na ospital. Sa edad na 12, ang mga convulsive seizure ay lumitaw nang walang pagkawala ng malay na may pagkibot ng kanang kamay, na kung saan ay isang serial na kalikasan, ang anticonvulsant therapy (Depakine) ay inireseta, ang dalas ng mga seizure ay nabawasan nang malaki. Sa edad na 13, ang isang MRI ng utak na may kaibahan ay ginanap - mga simetriko na pagbabago sa base ng temporal na lobes sa antas ng nuclei sa anyo ng isang pagtaas sa signal ng MR, na tipikal para sa nakakalason (mangganeso) o metabolic (tanso, bakal) encephalopathies.
Muling sinuri sa edad na 13 taon 3 buwan. isang endocrinologist, sa pag-aaral ng thyroid profile, ang pagtaas ng thyroid-stimulating hormone (TSH) ay nakita, ang subclinical hypothyroidism ay nasuri, at ang L-thyroxine ay inireseta.
Kapag pinag-aaralan ang card ng outpatient at dokumentasyon ng bata sa lugar ng paninirahan, ang pag-aaral ng calcium at phosphorus ay isinasagawa nang isang beses, sa edad na 1.5 taon, ang hypocalcemia ay nabanggit, ngunit walang karagdagang pagsusuri ang isinagawa sa okasyong ito. Dahil sa kawalan ng katiyakan ng diagnosis sa lugar ng paninirahan, ipinadala ng geneticist ang bata sa Moscow, sa Scientific Research Clinical Institute of Pediatrics, upang linawin ang diagnosis.
Layunin na datos ng pananaliksik:
Taas - 143 cm, timbang - 43 kg.
Ang pisikal na pag-unlad ay napakababa, maayos, ang pangangatawan ay hindi katimbang dahil sa pag-ikli ng mga limbs. Tumutugma ang Growth Sds sa -2.8 deviations mula sa norm (normal -2 + 2).
Mga tampok na phenotype: bilog na mukha, maikling leeg, anti-Mongoloid incision ng palpebral fissures, malawak na tulay ng ilong, mataas na noo, brachydactyly, pagpapaikli ng IV at V metacarpal at metatarsal bones (larawan 5). Sa mga panloob na organo - walang mga tampok. Sekswal na pag-unlad - Tanner III-IV yugto (na tumutugma sa edad).
Data mula sa laboratoryo at functional na pag-aaral:
Ang klinikal na pagsusuri ng dugo at ihi ay ang pamantayan.
Biochemical blood test: kabuuang calcium - 1.39 (normal 2.02-2.6 mmol / l), ionized calcium - 0.61 (normal 1.13-1.32 mmol / l), inorganic phosphorus - 3.66 (norm 0.86-1.56 mmol / l), iba pang mga tagapagpahiwatig ay nasa loob ang normal na hanay.
Biochemical analysis ng ihi: renal excretion ng phosphates ay nabawasan - 11.5 mmol / l (norm 19-32 mmol / l).
Profile ng thyroid: TSH - 11.75 (norm 0.4-4.0 μIU / ml), libreng T4 - 0.49 (norm 1.0-1.8 ng / dl).
Parathyroid hormone - 499 (normal 12-65 pg / ml), growth hormone - 7 ng / ml (normal 7-10 ng / ml), somatomedin-C - 250 ng / ml (normal 88-360 ng / ml).
Ultrasound ng mga panloob na organo - walang mga tampok.
ECG - paglipat ng supraventricular pacemaker laban sa background ng isang regular na rate ng puso na 71-80 beats / min. Hindi kumpletong pagbara sa kanang binti ng bundle ng Kanyang. Paglabag sa proseso ng repolarization sa myocardium ng posterior wall ng kaliwang ventricle (pagbaba sa z.T III, aVF).
R-graphy ng gulugod - right-sided scoliosis ng thoracic spine ng 1st degree, malubhang osteoporosis.
R-graphy ng mga kamay na may pagkuha ng mga bisig - pagpapaikli at pagpapalawak ng terminal at gitnang phalanges. Edad ng buto - 13.5–14 taon.
Ang mga pattern ng EEG ng aktibidad ng epileptik ay hindi nakarehistro.
MRI ng utak - MRI larawan ng maramihang subcortical foci ng tumaas na signal ng MR sa frontal lobes, panlabas na bayad na hydrocephalus na may pagkasayang ng sangkap ng utak.
MSCT ng utak - simetriko na lugar ng calcification ng lentiform nuclei. Nagkakalat ng mga hyperdense na lugar sa thalamus, caudate nuclei na may lugar ng calcification sa kanan. Maramihang mga tuldok na calcifications ng integumentary soft tissues ng bungo.
Audiogram - walang patolohiya.
Ang mga diagnostic ng DNA sa gene ng GNAS1 ay isinasagawa.
Payo ng eksperto:
Endocrinologist - Ang hereditary osteodystrophy type 1A ni Albright (pseudohypoparathyroidism). Pangunahing hypothyroidism, hindi kumpletong medikal na kabayaran.
Ophthalmologist - kumpletong pangalawang katarata. Inirerekomenda ang kirurhiko paggamot.
Psychologist - cognitive impairments (ang sikolohikal na profile ng bata ay ipinakita sa talahanayan).
Isinasaalang-alang ang phenotype ng bata, ang data ng kasaysayan, ang mga resulta ng mga karagdagang pag-aaral (hypocalcemia, hyperphosphatemia, hypophosphaturia, nadagdagan na parathyroid hormone ng dugo), mga calcification sa sangkap ng utak, ang pagkakaroon ng mga katarata, hypothyroidism), ang diagnosis ay ginawa: Albright's hereditary osteodystrophy type 1A (pseudohypoparathyroidism). Inirerekomenda na magsagawa ng mga diagnostic ng DNA - ang paghahanap para sa mga mutasyon sa gene ng GNAS1.
Paggamot: ang bata ay inirerekomenda na kumuha ng euthyrox sa isang dosis ng 100 mcg / araw; aktibong metabolite ng bitamina D - alpha-D3 ("Teva") sa isang dosis ng 2 mcg / araw; calcium ("Sandoz") 2000 mg / araw; patuloy na paggamit ng anticonvulsant therapy - finlepsin 800 mg / araw sa ilalim ng pangangasiwa ng isang neurologist-epileptologist; mga klase na may speech pathologist-defectologist at isang psychologist; energotropic therapy (Elkar at coenzyme Q10 sa mga dosis ng edad). Kontrol ng mga tagapagpahiwatig ng metabolismo ng phosphorus-calcium, mga antas ng parathyroid hormone.
Sa ganitong paraan, Ang ipinakita na klinikal na obserbasyon ay nagpapakita ng pagiging kumplikado ng pagkakaiba-iba ng diagnostic na paghahanap, ang kahalagahan ng napapanahong pag-aaral ng mga simpleng biochemical parameter (sa kaso ng epilepsy, ang paulit-ulit na screening ng mga tagapagpahiwatig ng phosphorus-calcium metabolism ay sapilitan), ang mga kinalabasan ng late diagnosis ng isang genetically determined disease. , ang pangangailangan na isama ang mga indibidwal na palatandaan sa pangkalahatang phenotype ng isang partikular na kondisyon ng pathological para sa naka-target na napapanahong pagsusuri ng ilang mga anyo ng mga namamana na sakit. Ang napapanahong pagsusuri, ang paglilinaw ng genesis ng bawat sindrom ay lalong mahalaga, dahil pinapayagan ka nitong mahanap ang pinakamahusay na diskarte sa paggamot ng mga kondisyong ito, ang pag-iwas sa mga posibleng komplikasyon (hanggang sa kapansanan ng bata); pag-iwas sa pag-ulit ng mga namamana na sakit sa mga apektadong pamilya (medical genetic counseling). Idinidikta nito ang pangangailangan para sa mga doktor ng iba't ibang mga specialty na malinaw na mag-navigate sa daloy ng namamana na patolohiya. Ang bibliograpiya ay nasa ilalim ng rebisyon.
Ang Albright's syndrome ay isang namamana na patolohiya na may hindi malinaw na pinagmulan. Tinatawag din itong pseudohypoparathyroidism. Ito ay nailalarawan sa pamamagitan ng pinsala sa tissue ng buto. Ang sakit ay nagpapakita ng sarili pangunahin sa babaeng kalahati ng populasyon, bagaman ito ay nangyayari rin sa mga lalaki. Halos lahat ng mga pasyente ay may mga paglihis sa mental at pisikal na pag-unlad. Ang mga buto ng tao ay nagsisimulang masira sa ilalim ng impluwensya ng mga metabolic na proseso ng posporus at kaltsyum. Nagsisimula ang paglabas ng parathyroid hormone ng parathyroid gland.
- Etiology
- Mga sintomas
- Mga diagnostic
- Paggamot
- Pag-iwas
Etiology
Ang pangunahing dahilan para sa naturang patolohiya bilang Forbes-Albright syndrome ay ang namamana na pagtutol ng mga tisyu ng katawan sa impluwensya ng parathyroid hormone. Kung ang isang tao ay walang sakit, kung gayon ang hormone ay kumikilos sa pamamagitan ng isang tiyak na sangkap. Kapag ang sakit ay nagpapakita mismo, ang komposisyon ng mga sangkap na ito ay nagbabago sa antas ng genetic. Kaya, ang mga peripheral tissue ay nagiging insensitive sa parathyroid hormone.
Mga sintomas
Ang Albright syndrome ay may tatlong pangunahing sintomas:
- Dahil sa mataas na antas ng hormonal, nangyayari ang maagang pagdadalaga. Ang mga batang babae ay maaaring magsimula ng regla sa unang sampung buwan ng buhay. Ngunit ang sintomas na ito ay hindi nalalapat sa mga lalaki. Nagsisimula sila sa pagbibinata gaya ng dati. Ang hormonal background ay nagdaragdag kapag ang mga malfunctions ay nangyari sa gawain ng adrenal glands, thyroid gland at pituitary gland. Ang sintomas na ito ay maaaring bumuo sa dalawang anyo - hindi kumpleto at kumpleto. Kung ang patolohiya ay nasa buong anyo, kung gayon ang regla ay nagsisimula nang maaga. Gayunpaman, ang mga ito ay masakit at sagana. Lumilitaw din ang mga pangalawang sekswal na katangian, halimbawa, ang mga glandula ng mammary ay tumaas. Kung ang anyo ng patolohiya ay hindi kumpleto, pagkatapos ay hindi magsisimula ang regla. Kadalasan, ang sakit na Albright sa mga batang babae ay sinamahan ng pag-unlad ng mga cyst sa mga ovary. Ang maagang pagdadalaga sa mga lalaki ay nailalarawan sa pamamagitan ng pagpapalaki ng ari ng lalaki at ang hitsura ng pubic hair.
- Fibrous osteodysplasia. Ito ay isang patolohiya kung saan ang tissue ng buto ay deformed, dahil sa kung saan ang mga buto ng balangkas ay baluktot. Lumilitaw ito bilang kawalaan ng simetrya. Ang mga visual na sintomas ay kurbada ng gulugod at pagkapilay. Ang patolohiya na ito ay nakakaapekto sa pangunahing mga tubular bones. Bumagal ang paglaki ng tao. Ito ay makikita kahit sa mga bata. Ang mga buto ng bungo ay umuunlad nang hindi pantay, lumilitaw ang mga nakaumbok na mata. Kapag natapos na ang pagdadalaga, hihinto ang pagbabago ng buto.
- Pagbabago sa kulay ng balat. Ang malakas na pigmentation ay nagsisimulang lumitaw. Lumilitaw ang dilaw-kayumanggi na mga spot na may hindi malinaw na mga gilid sa mga hita at sa katawan mismo. Ang mga spot ay malinaw na nakikita sa ibabang likod, itaas na likod, dibdib at pigi. Sa Albright's syndrome, ang mga spot sa katawan ng sanggol ay lilitaw kaagad pagkatapos ng kapanganakan.
Ang sakit na pseudohypoparathyroidism ay nagpapakita ng mga sintomas ng isang tiyak na kalikasan.
Mga diagnostic
Sa kaso ng pseudohypoparathyroidism, ang diagnosis ay ginawa batay sa isang visual na pagsusuri.
Ginagamit din ang mga pag-aaral sa laboratoryo at hardware:
- ang diagnosis ay ginawa kapag lumitaw ang dalawang pangunahing sintomas;
- isang konsultasyon sa isang geneticist ay naka-iskedyul;
- ang isang espesyal na x-ray ay ginanap;
- magsagawa ng pagsusuri sa dugo sa laboratoryo para sa mga hormone;
- kinakailangan ang konsultasyon ng isang pediatric endocrinologist;
- ang mga batang babae ay sinusuri ng isang pediatric gynecologist, at ang mga lalaki ay isang urologist;
- ang mga batang babae ay inireseta ng pagsusuri sa ultrasound ng mas mababang pelvic organ, at ang mga lalaki ay sumasailalim sa ultrasound ng scrotum;
- kung kinakailangan, ang mga karagdagang pagsusuri ng endocrine system ay inireseta.
Ang mga pasyenteng dumaranas ng Albright's syndrome ay dapat na regular na suriin ng mga espesyalista tulad ng endocrinologist, ophthalmologist, gynecologist, traumatologist.
Paggamot
Ang pseudohypoparathyroidism ay ginagamot nang komprehensibo.
Dahil ito ay isang genetic na patolohiya, ang therapy ay naglalayong alisin ang mga sintomas na lumitaw:
- Ang hormonal na paggamot ay inireseta. Makakatulong ito sa gawain ng endocrine system. Kinakailangan na maingat na subaybayan ang pagpapapangit ng mga buto ng bungo upang maiwasan ang pag-unlad ng patolohiya sa oras.
- Interbensyon sa pagpapatakbo. Ang pamamaraang ito ay ginagamit kapag ang pagpapapangit ng mga buto ng mukha ay maaaring humantong sa kapansanan sa pandinig at paningin.
- Magreseta ng mga painkiller na naglalaman ng bisphosphonates.
Para sa bawat pasyente na nagdurusa sa Albright's syndrome, ang paraan ng therapy ay pinili nang paisa-isa. Dapat na mahigpit na sundin ng pasyente ang lahat ng mga rekomendasyon at reseta ng doktor.
Ang pana-panahong pagsubaybay sa antas ng calcium sa katawan ng pasyente ay isinasagawa. Sa paunang yugto ng paggamot, ang naturang pag-aaral ay isinasagawa minsan sa isang linggo. Unti-unti, ang dalas ng mga sukat ay nababawasan sa isang beses sa isang buwan. Ito ay isasagawa hanggang sa katapusan ng iniresetang paggamot.
Kung ang pseudohypoparathyroidism ay nasuri, kung gayon ang pasyente ay dapat sumunod sa isang mahigpit na diyeta. Binubuo ito sa kumpletong pagbubukod mula sa menu ng mga produkto kung saan naroroon ang posporus.
Ang Albright syndrome (F. Albright) ay isang sakit na nailalarawan sa pamamagitan ng isang triad ng mga palatandaan: fibrous osteodysplasia, foci ng skin hyperpigmentation at precocious puberty. Mas karaniwan sa mga kababaihan.
Ang etiology ng Albright's syndrome ay hindi alam; nagmumungkahi ng mga neurogenic na impluwensya at mga pagbabago sa endocrine na dulot ng mga congenital na pagbabago sa hypothalamic region, na may napaaga na produksyon ng gonadotropic hormone ng anterior pituitary gland batay sa sclerosis at hyperostosis ng base ng bungo, pangunahin sa rehiyon ng Turkish saddle ( Fig.).
Ang paglaki at maturity ng skeleton (hitsura at pagsasanib ng ossification nuclei) ay maaaring higit na lumampas sa normal na mga halaga. Bilang resulta, ang mga carrier ng Albright's syndrome sa pagkabata ay maaaring mas matangkad, at pagkatapos ng napaaga na pagsasanib ng mga epiphyses, mas mababa kaysa sa malusog na mga kapantay. Ang pag-unlad ng balangkas ay nauuna sa wala sa panahon na pagdadalaga - kung minsan ay nabubura, bihirang makabuluhan, kapag ang pag-andar ng regla ay maaaring mangyari sa mga unang taon, at sporadic menorrhagia - kahit na sa mga unang buwan ng buhay. Ang maagang pagbibinata sa mga lalaki ay bihira.
Ang foci ng hyperpigmentation ng balat sa Albright's syndrome ay sinusunod sa puno ng kahoy, hita, mas madalas sa leeg, binti. Ang mga ito ay may hitsura ng malalaking patlang o magkahiwalay at nagsasama-samang mga spot na may diameter na 2 hanggang 20 cm o higit pa, hindi regular ang hugis na may tulis-tulis na mga gilid, o kinakatawan ng mga kumpol ng maputlang dilaw hanggang kayumangging mga pekas. Minsan ang mga foci na ito ay nakikita na sa kapanganakan, ngunit may maliit na sukat at pamumutla, maaari silang tingnan. Sa pamamagitan ng lokalisasyon, ang mga pagbabago sa balat ay hindi palaging nag-tutugma sa mga sugat sa buto. Ang mga hyperpigmented na bahagi ng balat ay hindi nakataas.
Sa Albright's syndrome, mayroong iba pang mga deviations - hyperthyroidism, diabetes, vascular defects, cartilaginous osteodysplasia, atbp. Ang mga biochemical parameter sa Albright's syndrome ay normal; kung minsan ang aktibidad ng alkaline phosphatase ay nadagdagan (hanggang sa 20 mga yunit ng Bodansky). Kabilang sa mga komplikasyon ng fibrous dysplasia, partikular para sa Albright's syndrome, mayroong pagkasayang ng optic nerve at exophthalmos bilang resulta ng hyperostosis ng sella turcica at orbit.
Ang pagbabala para sa Albright syndrome ay kanais-nais, ngunit ang mga pathological fracture at mga deformidad ng buto at, sa mga bihirang kaso, ang malignancy ng fibrous foci ay posible.
Differential diagnosis at paggamot ng bahagi ng buto ng Albright's syndrome - tingnan ang Osteodysplasia. Ang mga pagpapakita ng balat at endocrine sa Albright's syndrome ay hindi maibabalik.
Compaction ng istraktura ng buto ng base ng bungo, pangunahin sa rehiyon ng Turkish saddle.
Pseudohypoparathyroidism(Albright syndrome) ay isang sakit na nailalarawan sa pamamagitan ng mga pagbabago sa morphological, hypocalcemic tetany at kawalan ng tugon sa pagpapakilala ng parathyroid hormone. Sa sakit, maikli ang tangkad, matipunong pangangatawan, bilog na mukha, napaaga na pagsasara ng epiphyseal ossification nuclei, labis na katabaan, brachydactyly, minsan maiikling mga buto ng metatarsal, at mga dystrophic na pagbabago sa ngipin at mga kuko. Minsan mayroong isang lag sa katalinuhan, calcification ng malambot na mga tisyu. Hindi tulad ng totoong hypoparathyroidism, ang hypocalcemia, hyperphosphatemia at hypophosphaturia ay hindi naitama sa pamamagitan ng pangangasiwa ng parathyroid hormone.
Kadalasan ang mga taong nasa kabataan at may sapat na gulang ay nagkakasakit.
Iniuugnay ni Albright ang kondisyong ito sa refractoriness ng epithelium ng renal tubules sa pagkilos ng endogenous at exogenous parathyroid hormone, na humahantong sa pagtaas ng reabsorption ng phosphorus ng mga bato na may hyperphosphatemia, hypophosphaturia, hypercalcemia at hypercalciuria. Iniugnay niya ang pseudohypoparathyroidism sa iba pang mga katulad na estado ng organ refractoriness sa ilang mga hormone (tingnan ang Seabright-Bantam syndrome).
Sa sakit na ito, ang parathyroid gland ay may normal na istraktura.
Pseudopseudohypoparathyroidism- isang pathological na kondisyon na inilarawan ni Albright noong 1952, na kahawig ng pseudohypoparathyroidism na may likas na pagbabago sa morphological, ngunit walang katumbas na mga pagbabago sa humoral.
Mga karamdaman sa endocrine system
Kadalasan, ang mga batang babae na may Albright syndrome ay nakakaranas ng maagang pagbibinata, na sanhi ng mga estrogen na inilabas sa dugo mula sa mga ovarian cyst. Maaaring lumaki ang mga cyst, pagkatapos ay bumaba ang laki sa loob ng ilang linggo o araw. Sa tulong ng pamamaraan ng ultrasound, posible na makita at sukatin ang laki ng mga neoplasma. Ang mga cyst ay maaaring lumaki sa medyo disenteng laki. May mga kaso kapag ito ay lumaki sa laki ng isang bola ng golf, iyon ay, higit sa 50 mm ang lapad.
Ang paglaki ng dibdib at pagdurugo ng regla ay nakikita kasama ng paglaki ng cyst. Kung ang batang babae ay nagsimulang magkaroon ng regla bago ang edad na 2, kung gayon ito ang unang sintomas ng Albright's syndrome. Gayunpaman, ang pagkakaroon ng hindi regular na regla at mga ovarian cyst ay maaaring maobserbahan sa parehong mga kabataan at mga babaeng nasa hustong gulang. Ang lahat ng ito ay hindi nakakasagabal sa pagkakaroon ng malulusog na anak.
Ang paggamot sa mga bata na may maagang pagbibinata ay medyo mahirap at hindi epektibo. Kahit na ang cyst ay tinanggal sa pamamagitan ng operasyon, maaari itong muling lumitaw. Kapag kumukuha ng hormone progesterone, maaaring ihinto ang regla, ngunit ang mabilis na rate ng pag-unlad at paglaki ng mga buto ay hindi bumabagal. Mga posibleng negatibong kahihinatnan para sa gawain ng mga adrenal glandula. Ang paggamot ay may mga gamot sa bibig na humaharang sa estrogen synthesis.
Mga function ng thyroid
50 porsiyento ng mga taong may Albright syndrome ay dumaranas ng thyroid dysfunction. Ito ang tinatawag na goiter, nodules at cysts. Sa mga bihirang kaso, posible ang mga banayad na pagbabago sa istruktura. Sa mga pasyenteng ito, ang antas ng thyroid-stimulating hormone na ginawa ng pituitary gland ay mababa, ngunit ang mga antas ng thyroid hormone ay normal o bahagyang nakataas. Ang paggamot ay isinasagawa sa tulong kung saan ang synthesis ng mga thyroid hormone ay nabawasan. Ito ay ipinahiwatig sa mga kaso kung saan ang antas ng mga sikretong hormone ay sapat na mataas.

Labis na pagtatago ng growth hormone
Kapag may sakit, ang pituitary gland ay nagsisimulang mag-secrete ng malaking halaga ng growth hormone. Ang acromegaly ay natagpuan sa mga batang na-diagnose na may Albright's syndrome. Ang mga kabataang lalaki ay nagsimulang magkaroon ng magaspang na mga tampok ng mukha, ang kanilang mga braso at binti ay mabilis na tumaas, maaari silang magdusa mula sa arthritis. Ang paggamot sa mga bata na may ganitong mga palatandaan ay nabawasan sa operasyon ng pag-alis ng pituitary gland at ang paggamit ng mga synthesized analogues ng hormone somatostatin, na pinipigilan ang produksyon ng growth hormone.
Iba pang mga endocrine disorder
Bihirang sapat, mayroong labis na pagtatago at pagpapalawak ng mga adrenal glandula. Ang ganitong paglabag ay maaaring humantong sa labis na katabaan ng puno ng kahoy at mukha, pagtaas ng timbang, paghinto ng paglaki at pagkasira ng balat. Ang lahat ng mga sintomas na ito ay tinatawag na Cushing's syndrome. Sa ganitong mga pagbabago, ang apektadong adrenal gland ay aalisin o ang mga gamot ay iniinom na nagbabawas sa synthesis ng cortisol.
Minsan ang mga bata na may Albright syndrome ay may napakababang antas ng phosphorus sa dugo dahil sa malaking pagkawala ng phosphate sa ihi. Ang karamdaman na ito ay maaaring may pananagutan sa mga pagbabago sa buto na nauugnay sa mga ricket. Ang paggamot ay may mga oral phosphate at bitamina D bilang karagdagan.
Mga karamdaman sa balat
Ang mga café au lait spot ay lumilitaw sa balat mula sa kapanganakan o ilang sandali pagkatapos nito. Ang mga ito ay kadalasang nangyayari sa sacrum, trunk, limbs, pigi, likod ng leeg, noo, anit, batok. Ang lahat ng ito ay senyales din na may Albright syndrome ang bata. Ang mga larawan ng mga spot na ito ay makikita sa ibaba.

Bagama't may sakit tulad ng neurofibromatosis, mayroon ding mga café-au-lait spot. Gayunpaman, ang Albright's syndrome ay nailalarawan sa pamamagitan ng mas malalaking mga spot na may hindi regular na mga balangkas, mayroong mas kaunti sa kanila. Mayroon silang diameter na 1 hanggang ilang sentimetro, isang kayumangging kulay. Lahat sila ay may parehong kulay, ang mga ito ay hugis-itlog sa hugis, sila ay nailalarawan sa pamamagitan ng isang makinis na ibabaw. Ang mga pag-aaral sa kasaysayan ay kadalasang nagbubunyag na ang epidermis ay hindi nagbabago sa istraktura nito, ngunit ang dami ng melanin sa mga keratinocytes ay bahagyang tumaas.
Ang mga solong spot ng ganitong uri ay maaari ding mangyari sa ganap na malusog na mga tao. Kung hindi sila mag-abala at hindi lumalaki, kung gayon hindi na kailangan ng paggamot. Kung ang masinsinang paglago ay sinusunod, may mga spot ng hindi regular na hugis, pagkatapos ay inirerekomenda na suriin ang mga ito sa histologically. At pagkatapos ay inalis sa pamamagitan ng operasyon.

Konklusyon
Kaya, maaari nating sabihin na ang Albright's syndrome ay nailalarawan sa pamamagitan ng pinsala sa mga buto o bungo, ang pagkakaroon ng mga spot ng edad sa balat, at maagang pagdadalaga. Bagama't may mga pagkakataon na ang unang dalawang sintomas lamang ang naroroon. Sa pangkalahatan, ang pangunahing sintomas ng sindrom ay mga sugat sa buto (osteodysplasia). Gayunpaman, sa simula ng pagdadalaga, ang prosesong ito ay hihinto. Sa mga matatanda, ang mga pagbabago sa buto ay hindi umuunlad. Sa pangkalahatan, sa pagkakakilanlan at wastong paggamot, ang pagbabala para sa paggamot ng sakit na ito ay medyo kanais-nais.