Проявляющиеся признаки полностью обусловлены клеточным расположением мутировавшего гена. У некоторых пациентов диагноз подтверждается в раннем детстве, поскольку присутствуют явные аномалии в развитии костей и отмечается повышенная работа эндокринных желез (так, менструация может начаться в грудничковом возрасте – спорадические меноррагии).
Лейкоплакия – причины возникновения у детей и взрослых, диагностика, локализация и методы лечения
Энтеровирусная инфекция – возбудитель и пути передачи, симптомы, диагностика, методы лечения и профилактика
Синдром Дресслера – причины возникновения после инфаркта, симптомы, диагностика, методы лечения и профилактики
Болезнь Олбрайта характеризуется тремя главными признаками, при наличии двух из трех уже можно ставить этот диагноз:
- Преждевременное половое созревание. Симптом может проявляться в полной или неполной степени. Например, менструации начинаются очень рано, они обильны, болезненны, но при неполной форме симптома их может и не быть вовсе. В младенческом периоде появляются вторичные половые признаки, такие, как набухание и развитие молочных желез, часто появляются кисты на яичниках. У мальчиков же растут волосы на лобке и член.
- Фиброзная остеодисплазия. Рост скелета, его зрелость, окостенение значительно опережают стандартные показатели развития. Нарушение костной ткани ведет к искривлению костей, их деформации, характерна асимметричность. Самые частые признаки – хромота, искривление позвоночника. Рост замедляется, что заметно уже в детском возрасте, потом низкорослость начинает сопровождать еще и лишний вес. У многих больных происходит неравномерный рост черепа, возникает внешнее уродство, пучеглазие. В возрасте полового созревания изменения костей замедляются, количество переломов уменьшается.
- Изменения кожных покровов. Это проявляется в гиперпигментации. Кожный покров на туловище покрыт большими желто-коричневыми пятнами с неровными краями. Участки пигментации могут располагаться на пораженной стороне в области поясницы, на бедре, верхней части спины, ягодицах, груди. У ребенка светло-коричневые пятна появляются, как правило, сразу после рождения.
Синдром Олбрайта проявляется признаками поражения костной ткани, кожи и симптомами эндокринных расстройств.
- У больных развивается фиброзная остеодисплазия, проявляющаяся различными деформациями и скелетной кривизной. Чаще всего поражаются трубчатые кости на одной конечности, что приводит к хромоте и деформации позвоночника. Уже в раннем возрасте нарушается формирование скелета. У пациентов выявляется деформация челюсти и черепа: лицевые кости утолщаются, становятся широкими, грубыми и крупными. Так формируются внешние уродства: асимметричное лицо и односторонний экзофтальм. Затем появляются симптомы артрита. Фиброзно-кистозная дисплазия представляет собой фокальное замещение кортикального слоя кости пролиферирующими фибробластами. Образование кист приводит к деформации кости и возникновению переломов. Чаще всего встречается деформация бедра по типу «пастушьего посоха».
- На коже больных появляются участки гиперпигментации, представляющие собой скопления пятен неправильной формы с неравными краями. Окраска пятен часто варьируется от желтоватой до темно-коричневой. Очаги поражения локализуются на ногах, бедрах, туловище. Пятна имеют достаточно большие размеры и гладкую поверхность. Они присутствуют на коже с рождения или распространяются по мере роста ребенка. Эпидермис при этом не изменяется. Их интенсивный рост требует гистологического исследования и оперативного вмешательства.
- Эндокринные нарушения обусловлены дисфункцией структур гипоталамо-гипофизарной области и желез внутренней секреции, расположенных на периферии. Половое созревание начинается у полугодовалых детей и заканчивается уже в семь лет. У больных начинаются месячные, развиваются молочные железы, появляются вторичные половые признаки - рост груди и появление волос на лобке. Во время ультразвукового исследования яичников в них обнаруживают крупные фолликулярные кисты. При этом сами яичники остаются нормальных размеров.
- У больных наблюдаются тонические судороги, возникающие сами по себе или под воздействием эндогенных и экзогенных раздражителей - стресса, гипо- или гипертермии, чрезмерного перенапряжения.
- Кальцинаты в коже и мягких тканях часто воспаляются и изъязвляются, доставляя больным дискомфорт.
- Возникают остеохондромы, хондроматоз, субпериостальная резорбция мелких костей пальцев рук.
- Типичные внешние признаки - низкий рост, лунообразное лицо, избыточная масса тела, короткие пальцы.
- Избыток фосфора нарушает усвояемость магния, вызывая мигрень, аритмии, боли в спине.
- Нарушается конфигурация и подвижность суставов, формируются ложные суставы.
- К прочим признакам синдрома относятся: рвота, гематурия, помутнение хрусталика, частичное отсутствие зубной эмали.
- Неврологические нарушения обусловлены атрофией зрительного нерва, потерей слуха и зрения.
- Задержка психомоторного развития.
Выделяют атипичную или бессимптомную форму псевдогипопаратиреоза, при которой количество кальция и фосфора в крови остается нормальным, отсутствуют судороги и остеомаляция.
Причины синдрома Олбрайта
Синдром Олбрайта – это наследственная патология с неясным происхождением. Ее еще называют псевдогипопаратиреозом. Для него характерно поражение костной ткани. Заболевание проявляется в основном у женской половины населения, хотя встречается и у мальчиков. Практически у всех больных отмечаются отклонения в умственном и физическом развитии.
Кости человека начинают разрушаться под влиянием обменных процессов фосфора и кальция. Начинается выделение паратгормона паращитовидной железой.
Основная причина такой патологии, как синдром Форбса – Олбрайта – это наследственная устойчивость тканей организма к влиянию паратгормона. Если человек не болен, тогда гормон действует посредством определенного вещества. Когда заболевание проявляется, то состав этого веществ изменяется на генетическом уровне. Таким образом периферические ткани становятся нечувствительными к паратгормону.
Основные проявления
Все проявления этого заболевания можно разделить на несколько типов. Так, например, эндокринные проявления включают в себя:
- Низкорослость.
- Ожирение.
- Повышенное содержание сахара в крови.
- Лунообразное лицо.
- Олигоменорею.
В некоторых случаях псевдогипопаратитеоз сочетается с акромегалией, синдромом Кушинга, гинекомаститей.
Костно-суставные проявления патологии выражаются в:
- Укорочении размера пальцев рук.
- Экзостозах.
- Дисхондроплазии.
- Задержке прорезывания зубов.
- Гипоплазии зубной эмали.
- Подкожных оссификатах.
Симптомы псевдогипопаратиреоза выражаются и в нарушении нервной системы. При этом у детей с самого рождения могут проявляться судороги, а также выраженное отставание в умственном развитии.
Среди других симптомов нередко отмечаются мочекаменная болезнь, наличие крови в моче, рвота, катаракта.
Со стороны эндокринных нарушений возникают дисфункции гипофиза и периферических желез внутренней секреции.

Чаще всего встречаются нарушения полового развития, которые характеризуются ранним созреванием. Развивается преждевременное половое развитие, начиная с полугода и до семи летнего возраста, в зависимости от индивидуальных особенностей организма. Данная патология проявляется полной формой и неполной.
Полный вид отмечается наличием двух признаков: ранним наступлением месячных и развитием молочных желез. Во время неполной характерно проявление вторичных половых признаков без менструации. Так, могут проявляться ускоренный рост в высоту, однако при репродуктивном возрасте, такие девушки не имеют отличий от женщин с нормальным развитием.
Со стороны эндокринных нарушений возникают дисфункции гипофиза и периферических желез внутренней секреции
Если полная форма проходит с ускоренным развитием, то неполная форма по развитию удлиняется в несколько раз.
Синдром олбрайта отличается от истинного слишком раннего развития тем, что при гормональных дисфункциях не происходит полового волосяного роста, отмечается незначительное ускорение дифференцировки скелета.
В яичниках образовываются фолликулярные кисты, крупных размеров, но сами яичники не увеличиваются. Гонадотропные гормоны обычно остаются без изменений.
Также существует ряд эндокринных патологий, среди которых гиперпаратиреоз, гипертиреоз, акромегалия, синдром Иценко-Кушинга, рахит.
Возможна фиброзная остеодисплазия, которая проявляется изменением окостенения, различными деформациями, скелетной кривизной. Не редко при синдроме Олбрайта возникает поражение трубчатых костей с одной стороны скелета, что приводит к хромоте и деформации позвоночного столба – такое состояние можно увидеть на многочисленных фото в сети интернет.
Показатели роста и формирование скелета, во многих случаях, отличаются уже в раннем возрасте. У большего процента больных отмечается деформация челюстных костей и черепа, вследствие чего отмечается неправильный рост лица, который из-за гормональных изменений приводит к акромегалии (заболевание характеризуется утолщением кистей и лицевого участка черепа, а также они становятся слишком широкими) и прочим патологиям, проявляющимся во внешних уродствах.
Так, во время исследований, у детей с диагнозом акромегалия было отмечено появление слишком грубых черт лица, нижние и верхние конечности были сильно увеличены в размерах, кроме текущего заболевания, возникли симптомы артрита.
Со стороны кожных покровов отмечается гиперпигментация, которая появляется в форме скопления пятен или плоской неправильной формы, имеющие неравномерные края. Окрас пятен может быть желтоватого цвета или коричневого. Месторасположение гиперпигментации в основном на ногах, в области бедра или на туловище.
Факторы, способствующие развитию синдрома:
- врожденные аномалии гипоталамуса,
- психогенные факторы - стрессы, конфликты, неврозы,
- эндокринопатии,
- гиперостоз основания черепа,
- преждевременная секреция гонадотропинов.
Синдром Олбрайта - результат нарушенной работы паращитовидных желез. Их основной функцией является регуляция фосфорно-кальциевого обмена. При данной патологии железы плохо реагируют на колебания этих элементов или совсем не воспринимают их.
Генетически детерминированная устойчивость структурных элементов почек и скелета к паратгормону обусловлена поражением циторецепторов или дисфункцией аденилатциклазы. Мутация специфических генов приводит к подавлению продукции цАМФ, участвующей в метаболизме, происходящем под воздействием паратгормона.
Причины дефицита цАМФ различны:
- повреждение нуклеотидсвязывающих белков клеточных мембран,
- дефект циторецепторов эпителия,
- нарушение работы аденилатциклазы.
Эти патологические изменения делают периферические ткани интактными к гормону паращитовидных желез. Органы-мишени теряют чувствительность к паратгормону, что приводит к повышению в крови уровня фосфора и снижению концентрации кальция.
Пористость костей и кистообразование - характерные изменения, обнаруживаемые при данном синдроме. Кальций вымывается из костей и откладывается в виде кальцинатов в подкожно-жировой клетчатке, мышцах, внутренних органах, эндотелии артериальных сосудов. Со временем костная ткань замещается фиброзной. Скелетные аномалии нарушают мобильность больных и становятся причиной сильной боли.
Снижение витамина D в крови приводит к нарушению процесса всасывания кальция в кишечнике. Гипокальциемия стимулирует еще большее выделение в кровь паратгормона и усиленное вымывание кальция из костей, что приводит к развитию выраженной остеомаляции. Костные нарушения при этом заболевании считаются самыми тяжелыми.
На коже больных появляются пигментные пятна коричневого цвета с неровными краями. Эндокринные нарушения при данном недуге представлены ранним половым созреванием и появлением вторичных половых признаков.
Страшный диагноз под названием синдром Олбрайта имеет неясную этиологию.
Патология генетического происхождения проявляется повышенной пигментацией кожи и изменениями гормональной системы с ранним половым развитием. Редкое заболевание поражает весь организм человека.
Многие больные страдают не только от физической отсталости, пациентам при болезни Олбрайта свойственна еще и умственная неполноценность.
Псевдогипопаратиреоз – так еще называют патологию, получившую название по фамилии американского ученого Фуллера Олбрайта (Fuller Albright), открывшего это заболевание.
Частота болезни встречается во всем мире как один случай на сто тысяч или на один миллион человек.
Установить диагноз затруднительно, по этой причине болезнь Олбрайта может неверно распознаваться, что усложняет точное определение данных о ее распространенности.
Синдром Мак-Кьюна-Олбрайта (другой синоним патологии) развивается из-за генетических мутаций, которые начинают происходить в организме случайно, без обоснованных причин.
Паращитовидная железа организма, регулирующая уровень кальция и фосфора, выделяет паратгормон. У заболевшего таким синдромом человека резистентность тканей к этому гормону нарушена.
В итоге, костная система человека начинает разрушаться под действием результатов изменения метаболизма кальция и фосфора.
Для синдрома характерно появление на теле специфических пятен, фиброзной дисплазии и различных эндокринопатий. Среди последних самая частая – преждевременность полового развития.
Отдельные генетики утверждают о подавляющей частоте больных среди девочек, так как характерный симптом раннего полового созревания у девочек отмечен почти в 10 раз чаще. Прочие проявления синдрома встречаются у обоих полов практически одинаково.
Этиология синдрома неизвестна.
Предполагаются неврогенные влияния и эндокринные мутации, которые вызваны врожденными отклонениями в гипоталамической области при эмбриональном развитии и ранней продукцией гонадотропного гормона.
Ученые считают, что развитие синдрома Мак-Кьюна-Олбрайта обуславливается отсутствием реакции клеток почек и костей на действие гормонов паращитовидных желез.
Уровень кальция и фосфора в кровеносной системе регулируется паращитовидными железами, но при этом патологическом состоянии их интактное (равнодушное) реагирование к колебаниям указанных элементов приводит к возникновению гиперпаратиреоза (избыточной продукции паратгормона), изменению структуры костей.
При синдроме Мак-Кьюна-Олбрайта сбой вызван мутацией генов.
Нарушение может произойти в любом месте метаболической цепи: у кого-то поврежден рецептор, у ряда больных изменению подверглись белки мембраны клетки, у других наблюдается недостаточная продукция цАМФ.
Связывающие эти случаи генетическая обусловленность не передается по наследству: мутация происходит уже после оплодотворения.
Симптоматика
Проявляющиеся признаки полностью обусловлены клеточным расположением мутировавшего гена.
У некоторых пациентов диагноз подтверждается в раннем детстве, поскольку присутствуют явные аномалии в развитии костей и отмечается повышенная работа эндокринных желез (так, менструация может начаться в грудничковом возрасте – спорадические меноррагии).
У других больных симптомы проявляются позже. Основные патологические признаки сопровождаются общим отставанием в развитии человека – интеллектуальном и физическом.
Диагностика
Болезнь Олбрайта трудно диагностируется у младенца, а в возрасте 6-10 лет симптоматика проявляется уже выраженно. Наблюдаются множественные отклонения в развитии костной системы, снижен кальций в крови, повышены фосфор и паратгормон.
Симптоматика
Выделяют три основных признака заболевания. При наличии даже двух из трех можно ставить такой диагноз.
- Очень раннее половое созревание в результате сильно завышенного гормонального фона. Менструация может начаться у девочек в первые месяцы после рождения. У мальчиков, страдающих синдромом Олбрайта, очень редко наблюдается раннее половое созревание. Изменение в гормональном фоне происходит из-за неправильной работы надпочечников, гипофиза и щитовидной железы. Этот симптом может проявляться в полной или неполной форме. При полной форме менструации начинаются очень рано, обычно они обильные и болезненные. В младенческом возрасте при этом синдроме проявляются и вторичные половые признаки, например, набухание молочных желез. При неполном проявлении менструации не бывает. Очень часто при синдроме Олбрайта у девочек возникают кисты на яичниках. У мальчиков при раннем половом созревание наблюдается увеличение члена и оволосение лобка.
- Фиброзная остеодисплазия. Это заболевание говорит о нарушении костной ткани, приводит к искривлению и деформации костей. Характеризуется асимметричностью. Самые распространенные признаки – хромота и искривление позвоночника. Чаще всего подвергаются изменению трубчатые кости. Рост скелета замедляется, это заметно даже у детей. У многих пострадавших от синдрома Мак-Кьюна наблюдается неравномерный рост костей черепа, что приводит к внешнему уродству. При изменении лица возможно проявление пучеглазия. После достижения возраста полового созревания костные изменения замедляются, и частота переломов сокращается.
- Изменения кожных покровов. Проявляется в повышенной пигментации. Кожа на туловище и бедрах покрывается желто-коричневыми пятнами с неровными краями. Пигментацию можно увидеть на пояснице, верхней части спины, ягодицах и на груди. Пятна появляются сразу же после рождения ребенка.
Синдром Олбрайта – это наследственное заболевание, включающее комплекс патологий эндокринной системы, кожи и опорно-двигательного аппарата. Иногда эту болезнь называют псевдогипопаратиреоз, наследственная остеодистрофия или же «синдром яванской курицы».
В 1942 году данная патология была впервые задокументирована американским врачом Фуллером Олбрайтом, занимавшимся исследованием эндокринной системы, в особенности – метаболизмом кальция.
Синдром Олбрайта встречается очень редко, примерная частота заболевания – 7,9 на 1 млн человек, при этом патология чаще встречается у женского пола.
Любая наследственная патология обусловлена негативными мутациями в генах, из-за чего возникают нарушения развития какой-либо системы организма, связанной с функционированием этих генов.
Для болезни Олбрайта характерны мутации гена GNAS1, который кодирует специальный белок Gs – альфа. Этот белок напрямую связан с рецепторами паратиреоидного гормона (ПТГ).
Наследование остеодистрофии Олбрайта связано с Х-хромосомой, что доказывается отсутствием наследования от отца к сыну и высокой частотой встречаемости болезни у женского пола.
Снижение активности белка GS-альфа приводит к тому, что ткани организма становятся нечувствительны к паратгормону. Кроме этого, в клетках нарушается синтез цАМФ, молекулы, выполняющей функцию посредника для распространения сигналов некоторых гормонов.
Симптомы
Брахидактилия при синдроме Олбрайта Заболевание проявляется низкорослость Лунообразная форма лица
В зависимости от причины патологии, выделяют несколько типов болезни.
Псевдогипопаратиреоз, что характеризуется патологией клеточных рецепторов, связывающих паратгормон, выделяют в тип заболевания 1а. Для другого типа заболевания, 1b, характерно нарушение белков, связывающих рецепторы и ферменты между собой.
Для псевдогипопаратиреоза 2 типа характерен острый недостаток цАМФ.
Вне зависимости от того, какой тип заболевания, болезнь Олбрайта характеризуется определенной общей симптоматикой. Из-за нарушения развития эндокринной системы у больных может наблюдаться увеличение паращитовидных желез. Тело может развиваться непропорционально из-за укорочения нижних конечностей, что приводит к заметной вплоть до карликовости (см фото выше).
Пальцы рук тоже могут расти укороченными, что называется брахидактилией. Форма лица «лунообразная». Может возникать ожирение и остеопороз. Из-за нарушения развития опорно-двигательного аппарата может быстро развиться деформация позвоночника, появление ложных суставов и отсутствие подвижности в тех участках, где она должна быть естественным анатомическим образом.
Нередко у больных болезнью Олбрайта возникает синдром Иценко-Кушинга
Из-за неправильного развития костной ткани возникают проблемы с зубами, связанные с быстрым повреждением эмали. Из-за гипокальцемии существует высокий риск развития катаракты.
Для синдрома Олбрайта также характерная кальцификация мягких тканей из-за кальция, который высвободился из костей.
Скопления кальция могут образовываться в почках, различных мышцах, на стенках кровеносных сосудов и в миокарде, что способно привести к сердечным осложнениям.
В целом кальцинаты, даже находясь в подкожной клетчатке, способны поражать ткани вокруг себя, из-за чего образуются небольшие, но причиняющие дискомфорт язвочки.
Кроме вышеперечисленных изменений, на коже больного появляются пятна разного размера кофейного цвета (см. фото ниже).
Нарушение пигментации кожи (появляются коричневые пятна на теле похожие на родинку)
Отдельным моментом в списке неврологических нарушений являются тонические судороги. Они могут возникнуть как сами по себе, так и из-за действия каких-либо раздражителей, например, стресса, изнуряющей физической работы, резкого изменения температуры и т.д.
Диагностика
Так как псевдогипопаратиреоз наследственное заболевание, то при наличии в родословной людей с подобной патологией, особенно близких родственников, необходимо проконсультироваться у врача – генетика, чтобы оценить риск рождения больного ребенка.

У новорожденного достаточно сложно сразу же диагностировать синдром Олбрайта, но в детском возрасте (5-10 лет), это уже становится возможным, так как проявляются все симптомы, характерные для болезни.
Необходимо также провести ряд анализов, в частности, анализ крови, позволяющий определить уровень кальция, фосфора и паратгормона. По анализу мочи можно определить количество фосфора и цАМФ.
Существует ряд специальных диагностических тестов, направленных на изучение чувствительности ткани к паратгормону.
Рентгенодиагностика способна указать на изменения в скелете и Совокупность результатов анализов, физических и психических симптомов укажет, что это за болезнь и может ли это быть синдромом Олбрайта, так как похожая симптоматика может наблюдаться у таких наследственных заболеваний как синдром Шерешевского – Тернера.
Очень важна как можно более ранняя диагностика псевдогипопаратиреоз, что приводит к быстрому началу лечения. Чем более оперативно начато необходимое лечение, тем меньше риск возникновения аномалий умственного развития. Обычно больные синдромом Олбрайта наблюдаются у нескольких специалистов:
- гинеколог или уролог;
- рентгенолог;
- эндокринолог;
- ортопед;
- диетолог и т.д,
Это связано с комплексными симптомами патологии.
Практически всегда больным назначают разнообразные препараты кальция, так как, в отличие от больных с истинным гипопаратиреозом, инъекции паратгормона не приводят к нужному эффекту.
Очень важно подобрать нужную дозу для того, чтоб поддерживался нормальный гомеостаз.
Для более полноценного усвоения кальция вместе с препаратами на его основе, выписывают витаминные комплексы, особенно содержащие витамин D, например, таких как:
- Оксидевит;
- Кальцитриол;
- Дигидротахистерол.
Помимо приема лекарств, больным необходимо соблюдать строгую диету, тоже основанную на продуктах, богатых кальцием.
Нельзя допускать и обратного эффекта – превышения уровня кальция выше нормы, так как может возникнуть гиперкальциемия, сопровождающаяся упадком сил, головными болями, нарушениями со стороны желудочно-кишечного тракта.
В данном случае необходимо полностью отказаться от препаратов кальция и прежде всего вылечить гиперкальциемию.

Если же заболевание сопровождается значительными нарушениями эндокринной системы, то может потребоваться необходимость проведения гормональной терапии. Часто для гормональной терапии используют Тамоксифен – препарат, блокирующий эстрогенные рецепторы.
Уровень кальция проверяют каждую неделю для того, чтобы проверить, необходимо ли подкорректировать курс лечения, или нет. Впоследствии уровень кальция проверяют один раз в месяц, а впоследствии, при достижении благоприятных результатов, раз в квартал.
При судорогах вводят внутривенно 10% раствор хлорида или глюконата кальция. Очень важно вводить раствор медленно, так как существует риск интоксикации. Рекомендуется повторять внутривенные инъекции 2-3 раза в сутки.
Если наблюдаются какие-то специфические и конкретные нарушения, то их лечат соответствующие специалисты, например, катаракту – окулист, а заболевания зубов – стоматолог.
У взрослых патологические изменения костной ткани прогрессируют.
Хирургия
Хирургическое вмешательство при синдроме Олбрайта проводится тогда, когда аномалии развития костей способны повредить нерв.
Диагностика
Этиопатогенетические факторы
В основе псевдогипопаратиреоза лежит наследственная устойчивость тканей организма к влиянию парат-гормона. В обычных условиях последний действует через особого посредника - циклического аденозинмонофосфата (цАМФ). Именно он даёт клетке указание подчиниться регулирующему сигналу парат-гормона. При болезни Олбрайта на генетическом уровне изменяется структура посредника.
Болезнь Олбрайта передаётся следующим поколениям по интересной схеме, которая носит название Х-сцепленное доминантное наследование: патология от матери переходит к дочерям и сыновьям в 50 % случаев. От отца с такой же вероятностью дефектный ген наследуют только дочери. Встречаются семейные случаи заболевания.
Ген синдрома Олбрайта расположен в половой Х-хромосоме
Без регулирующего действия парат-гормона обмен кальция существенно смещается в сторону утилизации в костную ткань. В крови при этом наблюдается его недостаток, который приводит к таким явлениям:
- мышечные судороги;
- нарушение образования электрических импульсов в миокарде и изменение сердечного ритма;
- усиление выработки парат-гормона и увеличение паращитовидных желёз.
Избыток парат-гормона, в свою очередь, приводит к повышению уровня фосфора в крови.
Щитовидная и паращитовидные железы - основные регуляторы обмена кальция и фосфора в организме
Эта генетическая патология характеризуется аутосомно-доминантным типом наследования.
Основная причина – врожденный дефект, который делает периферические ткани нечувствительный к паратгормону.
В зависимости от того, как и где нарушена метаболическая цепочка, различают два типа синдрома.
Лечение болезни
Характерными видимыми признаками можно назвать:
- низкий рост;
- излишний вес;
- высокий уровень глюкозы;
- лунообразное лицо;
- нарушения полового созревания.
Могут проявляться и следующие симптомы:
- акромегалия;
- аутоиммунные заболевания;
- гинекомастия;
- болезнь Иценко-Кушинга и др.
Больные часто жалуются на кровь в моче, постоянную рвоту, изменения в зубах, судороги. При осмотре видна подкожная оссификация, при чем кальцинаты могут изъявляться.
Часто у больных укорочены пальцы, деформируются эпифизы трубчатых костей, могут быть признаки лентикулярной катаракты.
Почти всегда такие больные отстают в умственном развитии.
Псевдогипопаратиреоз лечится комплексно.
Так как это генетическая патология, то терапия будет направлена на устранение появившихся симптомов:
- Назначается гормональное лечение. Это поможет в работе эндокринной системы. Необходимо внимательно следить за деформацией костей черепа, чтобы вовремя предотвратить развитие патологии.
- Оперативное вмешательство. Такой способ применяют, когда деформация лицевых костей может привести к нарушению слуха и зрения.
- Назначают обезболивающие препараты с содержанием бисфосфонатов.
Для каждого больного, страдающего от синдрома Олбрайта, способ терапии подбирается в индивидуальном порядке. Пациент должен строго соблюдать все рекомендации и назначения врача.
Проводится периодический контроль уровня кальция в организме пациента. На начальном этапе лечения такое исследование проводится один раз в неделю. Постепенно частота замеров снижается до одного раза в месяц. Она будет проводиться до конца назначенного лечения.
Если диагностирован псевдогипопаратиреоз, тогда больному необходимо придерживаться строгой диеты. Она заключается в полном исключении из меню продуктов, в которых присутствует фосфор.
Наследственное заболевание чаще всего затрагивает организм целиком, приводя к множественным изменениям. Признаки патологии в этом случае чрезвычайно схожи с таковыми при гипопаратиреозе - дефиците выработки парат-гормона в околощитовидных железах.
Синдром Олбрайта - генетическое заболевание невыясненной этиологии, поражающее весь организм человека. Патология возникает сравнительно редко и проявляется асимметричной гиперпигментацией кожи, гормональным дисбалансом, полиостотической фиброзной дисплазией костей. Этот недуг относится к редким формам гонадотропнозависимого раннего полового созревания.
Синдром Олбрайта развивается в основном у представительниц прекрасной половины человечества. Он обусловлен спонтанной мутацией гена в процессе эмбриогенеза и не передается по наследству. Диагноз ставят детям возрасте 5-10 лет на основании признаков, характерных для данного заболевания.
Впервые клиника синдрома была описана в 1907 году ученым Олбрайтом, в честь которого он и получил свое название. Ученый наблюдал за низкорослыми и коренастыми девочками с круглым лицом и короткой шеей. У них были обнаружены мышечные спазмы, скелетные деформации, частичное отсутствие зубной эмали, отставание в умственном развитии, признаки ожирения.
Эндокринопатия проявлялась ранним половым созреванием: появлением менструаций, активным ростом грудных желез, появлением волос на лобке. Происхождение патологии Олбрайт объяснял резистентностью почечного эпителия к паратгормону, приводящей к ускоренному обратному всасыванию фосфора и развитию гиперфосфатемии.
При этом у больных паращитовидные железы имели нормальное строение, а гуморальные изменения отсутствовали.
В официальной медицине синдром получил название псевдогипопаратиреоз. Нарушения фосфорно-кальциевого обмена приводят к патологии костной системы. Подобные расстройства легко объяснить устойчивостью тканей к эндогенному паратгормону. У больных отмечается отставание в психофизическом развитии. При этом отсутствует реакция на введение экзогенного паратгормона.
Больным с синдромом Олбрайта требуется консультация специалистов в области гинекологии, эндокринологии, лучевой диагностики, офтальмологии, травматологии и ортопедии.
Данное заболевание в основном возникает у девочек.
Расстройства появляются вследствие мутации гена на эмбриональном этапе развития, не имеет способности передаваться наследственно. Проявляющиеся признаки полностью исходят от клеточного расположения, в которые входит мутировавший ген.
Диагностика
Отсутствие повышения резистентности почек к паратгормону является достаточным основанием для постановки диагноза «болезнь Олбрайта».
Проведение биохимического анализа крови, выявившего гипофосфатурию (недостаточное выведение фосфора с мочой), дополняется рентгенологическим исследованием для контроля изменения костей и мягких тканей.
Женщины обязательно проходят осмотр у гинеколога, ультразвуковое исследование таза и анализ на половой хроматин. Очаги пятен бывают видны при рождении, но из-за небольшого размера и бледности их часто не замечают.
Профилактика
Профилактика заключается в следующем:
- люди с такой патологией должны регулярно делать физические упражнения, чтобы укреплять мускулатуру;
- если женщина хочет забеременеть, тогда ей необходимо проконсультироваться с гинекологом и генетиком.
Такая патология диагностируется у одного человека на один миллион жителей планеты. К сожалению, на сегодняшний день еще нет специфической терапии. Существует лишь симптоматическое лечение. Но если пациент будет находиться под контролем врача, есть шанс на благоприятный исход.
Симптомы
- Из-за высокого гормонального уровня происходит раннее половое созревание. Месячные у девочек могут начаться в период первых десяти месяцев жизни. Но этот симптом не относится к мальчикам. У них половое созревание начинается как обычно. Гормональный фон увеличивается, когда происходят сбои в работе надпочечников, щитовидной железы и гипофиза. Развиваться этот симптом может в двух формах ‒ неполной и полной. Если патология полной формы, тогда месячные начинаются слишком рано. При этом они болезненные и обильные. Также появляются и вторичные половые признаки, например, увеличиваются молочные железы. Если форма патологии ‒ неполная, тогда месячные не начинаются. Часто болезнь Олбрайта у девочек сопровождается развитием кисты на яичниках. Раннее половое созревание мальчиков характеризуется увеличением полового органа и появлением волос на лобке.
- Фиброзная остеодисплазия. Это патология, при которой деформируется костная ткань, из-за чего искривляются кости скелета. Проявляется в виде асимметричности. Визуальные симптомы ‒ это искривление позвоночника и хромота. Такая патология поражает в основном трубчатые кости. Рост человека замедляется. Это можно заметить даже у ребенка. Неравномерно развиваются кости черепа, появляется пучеглазие. После того как закончится половое созревание, изменение костей приостановится.
- Изменение цвета кожи. Начинает появляться сильная пигментация. На бедрах и самом теле проступают желто-коричневые пятна с нечеткими краями. Пятна хорошо видны на пояснице, верхней части спины, груди и ягодицах. При синдроме Олбрайта пятна на теле ребенка появляются сразу после его рождения.
Заболевание псевдогипопаратиреоз проявляет симптомы специфического характера.
Врожденный вывих бедра – представляет собой одну из самых распространенных аномалий развития. Недоразвитие или дисплазия тазобедренного сустава носит как односторонний, так и двусторонний характер. Причины развития патологии до конца не изучены, но клиницистам известен широкий спектр предрасполагающих факторов, который может выступать в качестве провокатора недуга, начиная от генетической предрасположенности и заканчивая неадекватным протеканием беременности.
Остеохондропатия (совпадающих симптомов: 2 из 11)
Остеохондропатия - это собирательное понятие, включающее в себя заболевания, поражающие опорно-двигательную систему, на фоне происходит деформация и некроз пораженного сегмента. Примечательно то, что такие патологии наиболее часто встречаются у детей и подростков.
Нейрофиброма (совпадающих симптомов: 2 из 11)
Нейрофиброма - представляет собой доброкачественную опухоль, формирующуюся из нервных тканей как в центральной, так и в периферической нервной системе. В подавляющем большинстве ситуаций диагностируется у подростков и лиц среднего возраста.
Болезнь Шейермана-Мау (совпадающих симптомов: 2 из 11)
Болезнь Шейермана-Мау (син. кифоз Шейермана, дорсальный (дорзальный) ювенильный кифоз) - прогрессирующая деформация позвоночного столба, которая развивается во время активного роста организма. Без своевременной терапии может привести к серьезным последствиям.
Меланоз (совпадающих симптомов: 2 из 11)
Меланоз - большое количество накопленного в организме пигмента меланина. На определенных участках кожного покрова появляется сильная пигментация. Наиболее часто патологию диагностируют у женщин детородного возраста. Проблема вызывает дискомфорт, потому что видна визуально, лечение затруднительно. Любая начатая терапия принесет лишь небольшое облегчение, а в качестве профилактических мер часто советуют изменить образ жизни.
Источник
Диагностика

Синдром Олбрайта отличается не только резистентностью почек и скелета к паратгормону, но и других органов к соответствующим гормонам - яичников к гонадотропному, щитовидной железы к тиреотропному, надпочечников к антидиуретическому. Синдрому Олбрайта сопутствуют следующие эндокринопатии: гипертиреоз, акромегалия, синдром Иценко-Кушинга.
- Дисфункция щитовидной железы проявляется так называемым зобом, узелками и кистами органа.
- Избыточная продукция гипофизом гормона роста приводит к развитию акромегалии. Черты лица становятся грубыми, руки и ноги длинными, возникает артрит.
- Чрезмерная секреция гормонов надпочечников становится причиной ожирения, прекращения роста и кожной хрупкости - типичных признаков синдрома Кушинга.
Также отмечается повышенная частота аутоиммунных заболеваний, сахарного диабета, сосудистых пороков, хрящевой остеодисплазии.
К осложнениям синдрома Олбрайта относятся: атрофия зрительного нерва, экзофтальм, патологические переломы, деформация костей, озлокачествления фиброзных фокусов, кровоизлияния в различных частях организма, кровотечения, нарушение работы кишечника, печени, почек, пищеварительной системы.
Прогноз терапии при своевременном распознавании болезни Олбрайта относительно благоприятный, однако полностью излечить наследственный дефект ферментов невозможно. Нормализация содержания кальция и фосфора в организме позволит пациентам заниматься лёгким трудом, исключающим нервное перенапряжение и работу с машинами и механизмами.

При повторяющихся приступах мышечных судорог, плохо поддающихся медикаментозной коррекции, врачебной комиссией решается вопрос о присвоении группы инвалидности. При тяжёлом течении псевдогипопаратиреоза возможно развитие следующих осложнений:

Обмен кальция и фосфора в организме
Обмен веществ - важнейшая сторона жизнедеятельности организма человека. Каждую секунду в тканях и органах одни химические элементы превращаются в другие. Как правило, их содержание чётко регулируется. В особенности это касается электролитов - натрия, калия, кальция и фосфора. Именно благодаря их конкретным соотношениям возможна работа сердца и скелетных мышц, передача нервного электрического сигнала.
Кальций содержится во всех органах и тканях и играет несколько важнейших ролей:

Фосфор в большом количестве содержится в костной ткани и крайне необходим для нормальной работы головного мозга. В строго определённом соотношении кальций и фосфор присутствуют в крови. Избыток этих химических соединений выводится почками с мочой.
Парат-гормон - основной регулятор уровня кальция и фосфора в крови

От постоянного количества кальция и фосфора напрямую зависит благополучие организма. Процессом их обмена управляют щитовидная и паращитовидные железы:
- Первая вырабатывает гормон кальцитонин, благодаря которому кальций из крови переходит в состав костной ткани.
- Вторые при помощи парат-гормона ликвидируют нехватку кальция в крови путём вымывания его из костей.
Кроме того, обмен кальция и фосфора регулируют почки, выводя избыток этих химических соединений с мочой. Немалую роль в этом играет витамин Д.
Синдром Олбрайта - наследственное заболевание, обусловленное дефектом обмена кальция и фосфора в организме. Оно приводит к разнообразным изменениям костей, почек, пищеварительного тракта и психическим нарушениям.
Наследственная остеодистрофия впервые рассмотрена учёным Фуллером Олбрайтом в 1942 году. Заболевание в несколько раз чаще встречается среди женщин. В медицинской литературе описано всего около 300 случаев этой редкой патологии.
Классификация
Псевдогипопаратиреоз подразделяется на три основные разновидности:

Методы диагностики
Для распознавания болезни Олбрайта необходима совместная работа нескольких специалистов: уролога, невролога, психиатра, эндокринолога, генетика. Для установления верного диагноза используются следующие методы:

Дифференциальная диагностика проводится со следующими заболеваниями:

Псевдогипопаратиреоз требует пожизненного наблюдения врача-эндокринолога. В случае повторяющихся тяжёлых приступов судорог необходима госпитализация в профильное отделение стационара.
Лечение препаратами - основной компонент терапии псевдогипопаратиреоза. Главными задачами являются:
- нормализация уровня кальция и фосфора в крови;
- восстановление нормальной структуры костной ткани;
- профилактика приступов судорог;
- предупреждение образования камней в почках.
Для нормализации уровня кальция используют препараты Хлорид кальция, Глюконат кальция. Чаще всего эти медикаменты применяют в таблетированной форме. Однако для устранения мышечных судорог практикуется внутривенное введение фармакологических средств.
Глюконат кальция - медикамент, применяемый для лечения болезни Олбрайта
Нормализации уровня кальция и фосфора в крови способствуют препараты витамина Д:
- Кальций Д3;
- Оксидевит;
- Кальцитрин.
Кальций Д3 назначается при болезни Олбрайта
Назначение парат-гормона при псевдогипопаратиреозе неэффективно, поскольку ткани и органы нечувствительны к его влиянию.
Коррекция рациона
Важным компонентом терапии псевдогипопаратиреоза является коррекция рациона. При этом жизненно необходимы достаточная калорийность и дробное питание. Количество выпитой жидкости за сутки должно составлять не менее 1,5–2 л.
- бобы;
- зелёный горошек;
- чечевица;
- миндаль;
- кресс-салат;
- сельдерей;
- цветная капуста;
- укроп;
- базилик;
- молоко;
- йогурт;
- сметана.
Синдром Олбрайта – редкое заболевание, которое передает наследственным путем и проявляется поражением костной системы. Причиной этого становиться нарушение метаболизма кальция и фосфора. Он возникает из-за резистентности тканей к паратгормону, которые вырабатывают паращитовидные железы. Этот синдром часто сопровождает умственной и физической отсталостью в развитии.
Также этот симптом называют псевдогипопаратиреозом.
Впервые эту болезнь описывал ученый Олбрайт, в честь которого она и получила свое название.
Типы
Различают два типа псевдогипопаратиреоза в зависимости от уровня кальция в крови:
- Первый по клинической картине очень поход на идиопатический гипопаратиреоз – пониженный уровень кальция и нечувствительность к паратгормону.
- Второй показывает нормальный уровень кальция, поэтому называется псевдогипопаратиреозом.
Врачи отмечают, что одна форма болезни может перейти в другую. Иногда у членов семьи это заболевание может приобретать разные формы.
У мужчин эта патология вызывает нарушения репродуктивной функции, поэтому по мужской линии оно передается крайне редко.
Причины и развитие
Эта генетическая патология характеризуется аутосомно-доминантным типом наследования.
Основная причина – врожденный дефект, который делает периферические ткани нечувствительный к паратгормону.
В зависимости от того, как и где нарушена метаболическая цепочка, различают два типа синдрома.
Признаки
Характерными видимыми признаками можно назвать:
- низкий рост;
- излишний вес;
- высокий уровень глюкозы;
- лунообразное лицо;
- нарушения полового созревания.
Могут проявляться и следующие симптомы:
- акромегалия;
- аутоиммунные заболевания;
- гинекомастия;
- болезнь Иценко-Кушинга и др.
Больные часто жалуются на кровь в моче, постоянную рвоту, изменения в зубах, судороги. При осмотре видна подкожная , при чем кальцинаты могут изъявляться.
Часто у больных укорочены пальцы, деформируются эпифизы трубчатых костей, могут быть признаки лентикулярной катаракты.
Почти всегда такие больные отстают в умственном развитии.
Диагностика
Заболевание определяется у детей в возрасте от пяти до десяти лет. Врач получает характерную клиническую картину, выявляет большое количество аномалий в развитии скелета, наличие:
- гипокальцемии;
- гиперфосфатемии;
- уменьшенного выделения кальция и фосфора с мочой;
- повышенное содержание паратгормона;
- резистенция почечных канальцев и др.
Лечение
Лечение заключается в различных препаратах кальция, которые подбираются специально, чтобы поддерживать гомеостаз в нормальном состоянии.
Также обязательно введение витаминов группы D, кальцитриола, оксидевита.
Индивидуально разрабатывается диета, которая ограничивает продукты с высоким содержанием фосфора и способствует формированию нормального уровня кальция в крови.
Если есть сопутствующие заболевания эндокринной системы, назначают также ряд гормональных препаратов, в качестве заместительной терапии.
Судороги устраняют с помощью глюконата кальция или раствора хлористого кальция.
Если подходить к лечению комплексно и правильно, оно может дать вполне благоприятный результат. Но каждый такой человек, даже если он вылечился и ведет нормальный образ жизни, прежде чем завести ребенка, должен пройти обследование у генетика для выявления рисков.
Впервые в отечественной литературе данный синдром описал В. Р. Брайцев в 1922 году, определив его как «фиброзные опухоли». 15 лет спустя эндокринолог Фуллер Олбрайт сформулировал признаки этого системного заболевания так: «синдром, характеризующийся диссеминированным фиброзным оститом, полями пигментации и эндокринными расстройствами с преждевременным половым созреванием у девочек».
Иными словами, синдром Мак-Кьюна–Олбрайта–Брайцева – это системное заболевание, характеризующееся преждевременным половым развитием и костной патологией. Эта болезнь встречается преимущественно у представительниц женского пола и является врожденной наследственной патологией. До недавнего времени считалось, что это исключительно женская болезнь, однако потом были зафиксированы случаи заболеваний и среди мальчиков. Причиной развития синдрома считают мутацию в определенном гене.
Признаки синдрома Мак-Кьюна-Олбрайта-Брайцева
- Пигментные пятна. У всех больных присутствует особая пигментация неправильной формы цвета кофе с молоком. Пятна обычно располагаются на спине, груди, в области бедер, ягодиц и поясницы. Это своего рода маркер заболевания, пигментация присутствует с рождения или появляется по мере роста ребенка.
- Костная патология. Обычно поражаются длинные трубчатые кости (например, бедренные). Наличие полостей в костях приводит к их деформации, возникновению переломов и искривлению конечностей. Типичной является деформация бедра по типу «пастушьего посоха». Возможно поражение костей черепа. При этом возникают асимметрия лица, односторонний экзофтальм (пучеглазие). Поражение костей и частота переломов снижаются после завершения пубертатного периода.
- Преждевременное половое развитие. Оно может начаться с возраста 6-9 месяцев и до 7 лет. Чаще всего первые признаки полового созревания появляются у ребенка в 3 года. Может проявляться в полной и неполной формах. Полная форма у девочек характеризуется менархе (первая менструация) и увеличением молочных желез. Возможны длительные и обильные кровотечения, часто встречаются кисты на яичниках. При неполной форме менархе не наступает.
Преждевременное половое развитие у мальчиков встречается значительно реже, сопровождается симметричным увеличением яичек, затем ростом полового члена и оволосением на лобке, так же как при нормальном половом развитии.
- Ускорение костного возраста и ускоренное закрытие зон роста.
- Иногда наблюдаются неврологические и психические нарушения (задержка психического развития, судороги, атрофия зрительного нерва, потеря слуха).
- В литературе имеются сведения и о других эндокринных нарушениях при этом заболевании: акромегалия, гиперпаратиреоз (повышенная функция паращитовидных желез), гипертиреоз (повышенная функция щитовидной железы) и наличие узлов в щитовидной железе, гиперкортицизм (избыток глюкокортикоидных гормонов надпочечников), рахит, устойчивый к витамину D.
Диагностика синдрома Мак-Кьюна-Олбрайта-Брайцева
- Наличие характерных признаков.
- Консультация генетика.
- Рентгенологическое обследование.
- Половые гормоны и гонадотропины, тиреоидные гормоны, пролактин, другие тропные гормоны гипофиза (по показаниям).
- УЗИ органов малого таза для девочек и мошонки для мальчиков.
- Консультация эндокринолога и детского гинеколога для девочек или детского уролога для мальчиков.
- Обследование других органов эндокринной системы (по показаниям).
Лечение синдрома Мак-Кьюна-Олбрайта-Брайцева
Пациенты с данной патологией нуждаются в наблюдении гинеколога-эндокринолога, эндокринолога, врача лучевой диагностики, окулиста, ортопеда-травматолога. Помимо этого необходимо периодически определять уровень тропных и гонадотропных гормонов. Специфическое лечение данного синдрома не разработано. Чаще всего врачи избирают наблюдательную тактику. Девочкам терапия показана в случае длительного повышения уровня эстрогенов, сопровождающегося частыми и тяжелыми кровотечениями. По показаниям проводится коррекция костных деформаций. При развитии симптомов поражения других эндокринных желез лечение проводится врачами-эндокринологами.
Catad_tema Педиатрия - статьи
Псевдогипопаратиреоз (наследственная остеодистрофия Олбрайта): сложности дифференциально-диагностического поиска. Клиническое наблюдение
Е.В. Тозлиян, педиатр-эндокринолог, генетик, к. м. н., И.В. Шулякова, невролог, к. м. н.,
обособленное структурное подразделение «Научно-исследовательский клинический институт педиатрии» ГБОУ ВПО «Российский национальный исследовательский медицинский университет имени Н.И. Пирогова» Минздрава РФ, г. Москва
Ключевые слова:
дети, псевдогипопаратиреоз, наследственная остеодистрофия Олбрайта, ожирение, гипокальциемия, диагностика, резистентность к паратиреоидному гормону.
Keywords:
children, pseudohypoparathyroidism, Albright hereditary osteodystrophy, obesity, hypocalcemia, diagnostics, parathyroid hormone resistance.
Псевдогипопаратиреоз (греч. pseudes – ложный + гипопаратиреоз; синоним: наследственная остеодистрофия Олбрайта, синдром «яванской курицы») – редкое наследственное заболевание костной системы, имитирующее гипопаратиреоз и характеризующееся нарушением обмена кальция и фосфора; часто сопровождается задержкой умственного и физического развития. Заболевание описано впервые американским врачом-эндокринологом Albright F. в 1942 году . Распространенность заболевания составляет 7,9 на 1 млн человек .
ГЕНЕТИЧЕСКИЕ ДАННЫЕ
Псевдогипопаратиреоз (ПГП) – генетически гетерогенное заболевание. Данные о типе наследственной передачи противоречивы: как X-сцепленный доминантный , так и аутосомно-доминантный, аутосомно-рецессивный типы . В большинстве случаев развитие наследственной остеодистрофии Олбрайта связано с мутациями в расположенном на хромосоме 20 локусе 20q13 гена GNAS1 (Patten et al., 1990), кодирующего белок Gs-альфа, связанного с рецептором паратиреоидного гормона (ПТГ) . Подобный фенотип выявлен и у больных с интерстициальной делецией длинного плеча хромосомы 2 локуса 2q37 .
ПАТОГЕНЕЗ
В основе патогенеза псевдогипопаратиреоза лежит генетически обусловленная резистентность почек и скелета к действию парат-гормона в результате дефекта комплекса «специфический циторецептор – паратгормон – аденилатциклаза», что нарушает процесс образования в почках циклического 3"-, 5"-аденозинмонофосфата (цАМФ), являющегося внутриклеточным посредником действия паратгормона на метаболические процессы. Псевдогипопаратиреоз является генетически гетерогенным заболеванием. У части больных дефектен сам циторецептор, связывающий паратгормон (тип 1А псевдогипопаратиреоза), у других отмечается дефект нуклеотидсвязывающего белка, локализованного в липидном бислое клеточной мембраны и функционально связывающего рецептор с аденилатциклазой (тип 1B псевдогипопаратиреоза). У некоторых больных наблюдается ферментативная недостаточность самой аденилатциклазы (псевдогипопаратиреоз 2-го типа). Дефицит цАМФ, получающийся вследствие этих дефектов, ведет к нарушению синтеза специфических белков, определяющих биологический эффект паратгормона. Таким образом, теряется чувствительность органов-мишеней к паратгормону .
КЛИНИЧЕСКАЯ ХАРАКТЕРИСТИКА
В настоящее время выделяют 4 клинические формы патологии: типы 1А, 1В, 1С и 2. Знание их клинико-биохимических особенностей и данных генетических исследований позволяет провести дифференциальную диагностику в рамках самой нозологической формы.
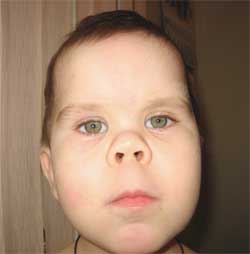
Общими признаками, позволяющими заподозрить заболевание, являются диспропорциональность физического развития, низкий рост (до карликовости) за счет укорочения нижних конечностей (фото 1), брахидактилия (фото 2), круглое «лунообразное» лицо (фото 3). Иногда наблюдаются экзостозы и аплазия зубов.
Фото 1.
Внешний вид ребенка с остеодистрофией Олбрайта
(особенности фенотипа, низкий рост за счет укорочения нижних конечностей)
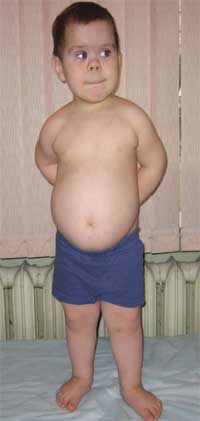
Фото 2.
Особенности костной системы у больного
с остеодистрофией Олбрайта
(брахидактилия – укорочение пальцев)
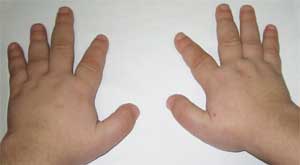
Фото 3.
Особенности фенотипа ребенка
с остеодистрофией Олбрайта
(круглое «лунообразное» лицо)
Патогномоничным признаком считается резкое укорочение I, III и V пястных и плюсневых костей (особенно III и IV), вследствие чего II пальцы на кистях и стопах оказываются длиннее остальных, а при сжатии кисти в кулак отсутствуют выпуклости в области IV и V пястно-фаланговых суставов – так называемый брахиметафалангизм. Выявляются также короткие широкие фаланги, утолщение свода черепа и деминерализация костей (остеопороз), ожирение .
Умственная отсталость (чаще умеренной степени выраженности) обнаруживается примерно у 20% больных. По данным некоторых авторов , олигофрения встречается в 70% случаев при наличии гипокальциемии и в 30% случае при нормокальциемии. Психические процессы у больных замедлены. В неврологическом статусе нередко отмечаются моторная неловкость, невротические реакции: страхи, тревога, беспокойство, плохой сон, повышение рефлексов, судороги, носящие тетанический характер и обусловленные гипокальциемией, иногда судорожные пароксизмы. Описаны также миопатические симптомы: мышечная утомляемость, мышечная слабость. Часто наблюдаются экстрапирамидные нарушения: хореиформные гиперкинезы, атетоз, лицевой гемиспазм, паркинсонизм, в отдельных случаях имеют место эпилептические пароксизмы, мозжечковые симптомы: атаксия, нарушение координации.
Нередко определяются кальцификация мягких тканей, подкожные кальцификаты (грудь, живот, пяточные сухожилия), при гистологическом исследовании которых – osteoma cutis (Izraeli et al., 1992), мозге (базальные ганглии). Важно отметить, что кальцификаты могут быть уже при рождении. Вследствие гипокальциемии обычно развивается катаракта и возникают дефекты эмали зубов.
ПСЕВДОГИПОПАРАТИРЕОЗ ТИПА 1А
имеет аутосомно-доминантный тип наследования. Ген псевдогипопаратиреоза типа 1А – GNAS1 – локализован на длинном плече хромосомы 20, в локусе 20q13.2. Развитие заболевания связано с дефицитом гуанин-нуклеотид-связывающего белка (Gs-белок). При этом ПТГ, связываясь с рецепторами тканей-мишеней, не способен активизировать циклический аденозинмонофосфат (цАМФ) и вызвать тканевой ответ. Вероятно, подобный механизм лежит в основе развития нечувствительности тканей других органов и эндокринных желез (гипофункция щитовидной железы, гонад, гипофиза, сахарный диабет, а также сниженный ответ печени на введение глюкагона), наблюдаемой при псевдогипопаратиреозе типа 1А. При данном типе патологии не наблюдается характерной для нормы повышенной экскреции цАМФ с мочой в ответ на экзогенное введение ПТГ. Заболевание диагностируется чаще в возрасте 5–10 лет. У больных наблюдаются низкий рост, короткая шея, круглое лицо, укорочение метакарпальных и метатарзальных костей (чаще укорочение IV и реже II пальцев) – так называемый брахи-метафалангизм. Отмечаются кальцификация мягких тканей, подкожные кальцификаты, которые могут выявляться уже при рождении; нередко наблюдается одновременное вовлечение других эндокринных желез: щитовидной железы (гипофункция), гонад, поджелудочной железы (сахарный диабет). Вследствие гипокальциемии нередко развиваются катаракта и дефект эмали зубов. В качестве дифференциально-диагностического теста отличия ПГП типа 1А от гипопаратиреоза: отсутствие клинического эффекта от парентерального введения ПТГ в виде подъема уровня кальция в крови и увеличения почечной экскреции фосфора с мочой (фосфатурический эффект).
При биохимическом исследовании выявляются гипокальциемия, гиперфосфатемия, увеличение уровня паратиреоидного гормона в крови, гипофосфатурия. Уровень Gs-белка в крови снижен. При рентгенологическом исследовании костной системы обнаруживаются укорочение метакарпальных и метатарзальных костей, генерализованная деминерализация, утолщение костей свода черепа.
ПСЕВДОГИПОПАРАТИРЕОЗ ТИПА 1B
имеет аутосомно-доминантный тип наследования, однако не исключен доминантный, сцепленный с Х-хромосомой тип наследования. Необходимо иметь в виду наблюдающуюся иногда неполную пенетрантность гена болезни и возможность скрытого носительства патологии. Поэтому рекомендуется клиническое (выявление субклинического течения болезни) и биохимическое обследование (определение уровней кальция, фосфора, ПТГ крови) предполагаемых носителей заболевания. ПГП типа 1В обусловлен дефицитом тканевых рецепторов к паратиреоидному гормону в органах-мишенях и ограниченной резистентностью к паратгормону. Клиническая картина сходна с клиникой типа 1А, но отсутствует поражение других эндокринных желез, реже встречается остеодистрофия.
У больных отсутствует реакция почек на экзогенное введение паратиреоидного гормона в виде увеличения экскреции циклического аденозинмонофосфата с мочой, однако, в отличие от типа 1А, уровень Gs-белка в крови нормален. Женщины поражаются чаще мужчин, однако тяжесть заболевания может быть одинаковой как у мужчин, так и у женщин.
ПСЕВДОГИПОПАРАТИРЕОЗ ТИПА 1С
некоторые авторы отождествляют с псевдо-псевдогипопаратиреозом (ППГП), описанным Albright F. в 1952 году. Характеризуется свойственной ПГП клинической картиной, однако уровни кальция, фосфора в крови и моче остаются в пределах нормы. Показатели ПТГ и Gs-белка в крови также сохраняются на нормальном уровне. У некоторых больных с ПГП типа 1С обнаруживаются делеции de novo на хромосоме 2 . Не исключено, что этот вариант болезни является подтипом ПГП типа 1А.
ПСЕВДОГИПОПАРАТИРЕОЗ ТИПА 2
клинически сходен с другими типами заболевания, однако имеет аутосомно-рецессивный тип наследования. Не исключено существование и аутосомно-доминантных форм патологии. Патогенез развития связан с внутриклеточной резистентностью к цАМФ. ПТГ при этом связывается с рецепторами и вызывает нормальную ответную реакцию клеток на ПТГ в виде увеличения экскреции цАМФ. Внутриклеточная нечувствительность к цАМФ, однако, не позволяет осуществиться полной реализации действия ПТГ. При этом сохраняется нормальная реакция почек на экзогенное введение паратиреоидного гормона в виде увеличения экскреции циклического аденозинмонофосфата с мочой. Высказывается мнение, что ПГП типа 2 может быть связан с дефицитом витамина D .
Таким образом, выделенные типы ПГП клинически характеризуются пониженной чувствительностью органов-мишеней к ПТГ, однако различаются патогенетическими механизмами формирования нечувствительности тканей.
ДИАГНОСТИКА
Лабораторным дифференциально-диагностическим тестом может служить характер почечной экскреции цАМФ в ответ на введение ПТГ: повышенная экскреция цАМФ отмечается при типе 2 и ее отсутствие – при типе 1. Диагноз подтверждается обнаружением сниженного уровня гуанин-нуклеотидсвязывающего белка (Gs-белок) в крови (в среднем в 1,5–2 раза) по сравнению с нормой. Гипокальциемия, как правило, сочетается с гиперфосфатемией и гипофосфатурией. Уровень ПТГ повышен; при 1C-типе уровень ПТГ в норме, что дало основание для названия «псевдогипопаратиреоз». При рентгенологическом исследовании костной системы обнаруживаются укорочение пястных и плюсневых костей, нередко генерализованная деминерализация (остеопороз), утолщение костей свода черепа. В дерматоглифическом рисунке отмечается смещение осевого ладонного трирадиуса.
Критерии диагноза:
- низкий рост;
- круглое лицо;
- задержка нервно-психического развития;
- скелетные аномалии;
- низкое содержание кальция в сыворотке крови;
- высокий уровень паратиреоидного гормона в крови;
- снижение экскреции с мочой фосфатов и цАМФ.
ЛЕЧЕНИЕ И ПРОФИЛАКТИКА
Лечение при гипокальциемии заключается в назначении препаратов кальция в дозах, достаточных для поддержания нормальной концентрации кальция в крови. Большое значение имеет терапия витамином D. В настоящее время применяют активные метаболиты витамина D – оксидевит, 1-альфа-Д3, кальцитрин и др. в дозе 1–2 мкг/сутки с положительным результатом (увеличение содержание кальция в крови, уменьшение проявлений судорожного синдрома). Эффективен также тахистин (0,5–1,5 мг/сутки). Данный препарат увеличивает всасывание кальция в кишечнике и тем самым способствует повышению уровня кальция в крови. Противосудорожная терапия используется как дополнительное лечение. На интеллектуальное развитие лечение не оказывает заметного действия, но наряду с уменьшением симптомов судорожного синдрома наблюдается регресс неврологических проявлений (подкорковых нарушений, хореиформных гиперкинезов, атетоза и др.). Во избежание передозировки препаратов витамина D необходим контроль концентрации кальция в крови каждые 3–7 дней в течение первых 2 недель лечения и каждый месяц в течение последующих 2–3 месяцев. По достижении стабильной концентрации кальция в крови достаточно проверять ее 1 раз в 2–3 месяца. Диета с ограничением фосфора помогает нормализовать как концентрацию фосфора, так и содержание кальция в крови и устранить симптомы вторичного гиперпаратиреоза. При недостаточности других желез внутренней секреции проводят заместительную терапию соответствующими гормонами.
Лечение паратгормоном неэффективно. Для купирования судорожных приступов внутривенно вводят 10%-ный раствор кальция хлорида или кальция глюконата; внутрь – 5–10%-ный раствор кальция хлорида по 1 столовой ложке 3–4 раза в день: кальция глюконат, кальция лактат – до 10 г в день.
ПРОГНОЗ для жизни определяется выраженностью судорожного синдрома.
ПРОФИЛАКТИКА болезни основывается на данных медико-генетического консультирования.
МЕДИКО-ГЕНЕТИЧЕСКОЕ КОНСУЛЬТИРОВАНИЕ
При медико-генетическом консультировании следует исходить из аутосомно-доминантного типа наследования и высокого (50%) риска повторения заболевания в семье при унаследованных формах. С целью идентификации характера типа наследования необходимо проводить тщательное обследование родителей, так как синдром может проявляться минимальными клиническими симптомами. В настоящее время разработана и совершенствуется молекулярно-генетическая диагностика заболевания путем типирования мутаций в гене GNAS1 на хромосоме 20. Разрабатываются способы пренатальной диагностики заболевания в целом и отдельных его типов.
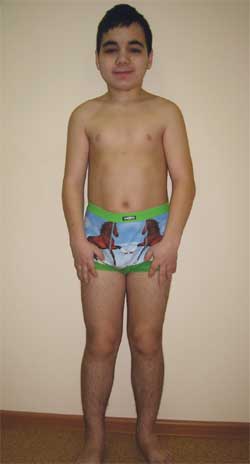
КЛИНИЧЕСКОЕ НАБЛЮДЕНИЕ Мальчик Г., 14,5 лет (фото 4), поступил в Научно-исследовательский клинический институт педиатрии с диагнозом: дегенеративное заболевание нервной системы? врожденная наружная гидроцефалия; симптоматическая эпилепсия; наследственный синдром? болезнь накопления? метаболическая энцефалопатия; субклинический гипотиреоз; низкорослость смешанного генеза; когнитивные нарушения.
Жалобы при поступлении на интенсивные приступообразные головные боли, локализующиеся в лобной области и сопровождающиеся рвотой, которая приносит облегчение, снижение памяти и успеваемости в школе, судорожные приступы, во время которых происходит подергивания в правой руке.
Фото 4.
Ребенок Г., 14,5 лет, с остеодистрофией Олбрайта
(особенности фенотипа, низкий рост, укорочение конечностей, брахидактилия)
Анамнез семейный: родители – армяне по национальности, не состоящие в кровном родстве и не имеющие профессиональных вредностей. В родословной случаев психических заболеваний, эпилепсии, задержки в развитии – не отмечалось. Сибс, сестра 17 лет – со слов – здорова.
Анамнез жизни и заболевания:
мальчик от 2-й беременности, протекавшей без особенностей, роды вторые, в срок, физиологические, масса при рождении – 3100 г, длина – 51 см. Закричал сразу, оценка по шкале Апгар – 7/9 баллов. Ухудшение состояния на 3-и сутки – судороги неонатальные, купированы в роддоме. Ранний постнатальный период – без особенностей. Отмечалась незначительная темповая задержка моторного развития на первом году жизни, самостоятельная ходьба с 1 года 3 мес. В связи с чем наблюдался неврологом с диагнозом: органическое поражение ЦНС; врожденная гидроцефалия; неонатальные судороги; фебрильные судороги в анамнезе.
Получал диакарб, финлепсин. Дебют приступов с 1 года 11 мес. – асимметричные, тонические в виде напряжения правой руки и ноги, с заведением глаз, до 2 мин., без потери сознания, частые до 10 эпизодов в сутки. Получал депакин нерегулярно. На фоне самостоятельной отмены – однократный тонический статус. В 2 года проведена по месту жительства КТ головного мозга, где выявлены единичные очаги демиелинизации в затылочных долях.
Консультирован нейрохирургом, рекомендовано консервативное лечение. С 3 лет отмечается задержка психоречевого развития, рекомендовано наблюдение психиатра.
С 4–5 лет родители стали отмечать деформацию и укорочение пальцев стоп и кистей, в особенности II–IV пальцев симметрично на руках и ногах, снижение ростовых показателей. В 8 лет заключение логопеда – общее нарушение речи 2–3-го уровня, рекомендовано обучение в специализированной школе. В этом же возрасте осмотр генетиком по месту жительства, заключение: наследственная болезнь обмена? рекомендовано исследование аминокислот крови, изменений не было выявлено; окончательное заключение: данных за наследственное заболевание обмена не выявлено; гипохондроплазия; рекомендовано лечение невролога и эндокринолога.
Таблица.
Профиль психического развития ребенка Г., 14,5 лет (IQ = 68)

В возрасте 8 лет консультирован эндокринологом по поводу задержки роста и развития. При рентгенологическом исследовании кистей рук отмечены особенности: средние, основные фаланги и пястные кости укорочены, утолщены; диагноз рентгенолога – ахондроплазия.
Неоднократно обследуется по месту жительства в неврологическом стационаре. В 12 лет появились судорожные приступы без потери сознания с подергиванием правой руки, носящие серийный характер, назначена противосудорожная терапия (депакин), частота приступов значительно сократилась. В 13 лет проведена МРТ головного мозга с контрастированием – симметричные изменения в основании височных долей на уровне ядер в виде повышения МР-сигнала, что характерно для токсических (марганец) или метаболических (медь, железо) энцефалопатий.
Вновь осмотрен в возрасте 13 лет 3 мес. эндокринологом, при исследовании тиреоидного профиля выявлено повышение тиреотропного гормона (ТТГ), диагностирован субклинический гипотиреоз, назначен L-тироксин.
При анализе амбулаторной карты ребенка и документации по месту жительства исследование кальция и фосфора проводилось однократно, в возрасте 1,5 лет, отмечалась гипокальциемия, но по данному поводу дообследование не проводилось. Учитывая неопределенность диагноза по месту жительства, генетиком ребенок направлен в Москву, в Научно-исследовательский клинический институт педиатрии, с целью уточнения диагноза.
Данные объективного исследования:
Рост – 143 см, масса – 43 кг.
Физическое развитие очень низкое, гармоничное, телосложение диспропорциональное за счет укорочения конечностей. Sds роста соответствует –2,8 отклонениям от нормы (норма –2+2).
Особенности фенотипа: круглое лицо, короткая шея, антимонголоидный разрез глазных щелей, широкое переносье, высокий лоб, брахидактилия, укорочение IV и V пястных и плюсневых костей (фото 5). По внутренним органам – без особенностей. Половое развитие – Tanner III–IV стадия (что соответствует возрасту).
Данные лабораторных и функциональных исследований:
Клинический анализ крови и мочи – норма.
Биохимический анализ крови: общий кальций – 1,39 (норма 2,02–2,6 ммоль/л), кальций ионизированный – 0,61 (норма 1,13–1,32 ммоль/л), фосфор неорганический – 3,66 (норма 0,86–1,56 ммоль/л), остальные показатели в пределах нормы.
Биохимический анализ мочи: почечная экскреция фосфатов снижена – 11,5 ммоль/л (норма 19–32 ммоль/л).
Тиреоидный профиль: ТТГ – 11,75 (норма 0,4–4,0 мкМЕ/мл), свободный Т4 – 0,49 (норма 1,0–1,8 нг/дл).
Паратиреоидный гормон – 499 (норма 12– 65 пг/мл), СТГ – 7 нг/мл (норма 7–10 нг/мл), соматомедин-С – 250 нг/мл (норма 88–360 нг/мл).
УЗИ внутренних органов – без особенностей.
ЭКГ – миграция суправентрикулярного водителя ритма на фоне регулярной ЧСС 71– 80 уд/мин. Неполная блокада правой ножки пучка Гиса. Нарушение процесса реполяризации в миокарде задней стенки левого желудочка (снижение з.Т III, аVF).
R-графия позвоночника – правосторонний сколиоз грудного отдела позвоночника 1-й степени, выраженный остеопороз.
R-графия кистей рук с захватом предплечий – укорочение и расширение концевых и средних фаланг. Костный возраст – 13,5– 14 лет.
ЭЭГ-паттернов эпилептической активности не зарегистрировано.
МРТ головного мозга – МР-картина множественных субкортикальных очагов повышенного МР-сигнала в лобных долях, наружная компенсированная гидроцефалия с атрофией вещества головного мозга.
МСКТ головного мозга – симметричные участки обызвествления лентиформных ядер. Диффузные гиперденсивные участки в таламусах, хвостатых ядрах с участком обызвествления справа. Множественные точечные обызвествления покровных мягких тканей черепа.
Аудиограмма – без патологии.
ДНК-диагностика в гене GNAS1 – в работе.
Консультации специалистов:
Эндокринолог – наследственная остеодис-трофия Олбрайта типа 1А (псевдогипопара-тиреоз). Первичный гипотиреоз, неполная медикаментозная компенсация.
Окулист – катаракта полная вторичная. Рекомендовано оперативное лечение.
Психолог – когнитивные нарушения (психологический профиль ребенка представлен в табл.).
Учитывая фенотип ребенка, данные анамнеза, результаты дополнительных исследований (гипокальциемия, гиперфосфатемия, гипофосфатурия, повышение паратиреоидного гормона крови), кальцинаты в веществе головного мозга, наличие катаракты, гипотиреоза), поставлен диагноз: наследственная остеодистрофия Олбрайта 1А-типа (псевдогипопаратиреоз). Рекомендовано проведение ДНК-диагностики - поиск мутаций в гене GNAS1.
Лечение: ребенку рекомендован прием эутирокса в дозе 100 мкг/сутки; активный метаболит витамина Д - альфа-Д3 («Тева») в дозе 2 мкг/сутки; кальций («Сандоз») 2000 мг/сутки; постоянный прием противосудорожной терапии - финлепсин 800 мг/сутки под наблюдением невролога-эпилептолога; занятия с логопедом-дефектологом и психологом; энерготропная терапия (Элькар и коэнзим Q10 в возрастных дозах). Контроль показателей фосфорно-кальциевого обмена, уровня паратгормона.
Таким образом,
представленное клиническое наблюдение демонстрирует сложности дифференциально-диагностического поиска, важность своевременного исследования простых биохимических параметров (при эпилепсии обязателен неоднократный скрининг показателей фосфорно-кальциевого обмена), исходы поздней диагностики генетически детерминированного заболевания, необходимость интегрировать отдельные признаки в общий фенотип того или иного патологического состояния для целенаправленной своевременной диагностики отдельных форм наследственных заболеваний. Своевременная диагностика, уточнение генеза каждого синдрома особенно важны, так как позволяют найти оптимальный подход к лечению этих состояний, профилактике возможных осложнений (вплоть до инвалидности ребенка); предупреждение повторного возникновения наследственных болезней в пораженных семьях (медико-генетическое консультирование). Это диктует необходимость врачам различных специальностей четко ориентироваться в потоке наследственно обусловленной патологии. Список литературы находится в редакции.
Синдром Олбрайта – это наследственная патология с неясным происхождением. Ее еще называют псевдогипопаратиреозом. Для него характерно поражение костной ткани. Заболевание проявляется в основном у женской половины населения, хотя встречается и у мальчиков. Практически у всех больных отмечаются отклонения в умственном и физическом развитии. Кости человека начинают разрушаться под влиянием обменных процессов фосфора и кальция. Начинается выделение паратгормона паращитовидной железой.
- Этиология
- Симптоматика
- Диагностика
- Лечение
- Профилактика
Этиология
Основная причина такой патологии, как синдром Форбса – Олбрайта – это наследственная устойчивость тканей организма к влиянию паратгормона. Если человек не болен, тогда гормон действует посредством определенного вещества. Когда заболевание проявляется, то состав этого веществ изменяется на генетическом уровне. Таким образом периферические ткани становятся нечувствительными к паратгормону.
Симптоматика
Синдром Олбрайта имеет три основных симптома:
- Из-за высокого гормонального уровня происходит раннее половое созревание. Месячные у девочек могут начаться в период первых десяти месяцев жизни. Но этот симптом не относится к мальчикам. У них половое созревание начинается как обычно. Гормональный фон увеличивается, когда происходят сбои в работе надпочечников, щитовидной железы и гипофиза. Развиваться этот симптом может в двух формах ‒ неполной и полной. Если патология полной формы, тогда месячные начинаются слишком рано. При этом они болезненные и обильные. Также появляются и вторичные половые признаки, например, увеличиваются молочные железы. Если форма патологии ‒ неполная, тогда месячные не начинаются. Часто болезнь Олбрайта у девочек сопровождается развитием кисты на яичниках. Раннее половое созревание мальчиков характеризуется увеличением полового органа и появлением волос на лобке.
- Фиброзная остеодисплазия. Это патология, при которой деформируется костная ткань, из-за чего искривляются кости скелета. Проявляется в виде асимметричности. Визуальные симптомы ‒ это искривление позвоночника и хромота. Такая патология поражает в основном трубчатые кости. Рост человека замедляется. Это можно заметить даже у ребенка. Неравномерно развиваются кости черепа, появляется пучеглазие. После того как закончится половое созревание, изменение костей приостановится.
- Изменение цвета кожи. Начинает появляться сильная пигментация. На бедрах и самом теле проступают желто-коричневые пятна с нечеткими краями. Пятна хорошо видны на пояснице, верхней части спины, груди и ягодицах. При синдроме Олбрайта пятна на теле ребенка появляются сразу после его рождения.
Заболевание псевдогипопаратиреоз проявляет симптомы специфического характера.
Диагностика
При заболевании псевдогипопаратиреозом проводится диагностика, основанная на визуальном осмотре.
Также применяются лабораторные и аппаратные исследования:
- диагноз ставят при появлении двух основных симптомов;
- назначается консультация генетика;
- проводится специальный рентген;
- проводят лабораторный анализ крови на гормоны;
- требуется консультация детского эндокринолога;
- девочек осматривает детский гинеколог, а мальчиков ‒ уролог;
- девочкам назначают ультразвуковое обследование органов нижнего таза, а мальчикам проводят УЗИ мошонки;
- при необходимости назначают дополнительные обследования эндокринной системы.
Больные, страдающие от синдрома Олбрайта, должны регулярно проходить осмотры у таких специалистов, как эндокринолог, окулист, гинеколог, травматолог.
Лечение
Псевдогипопаратиреоз лечится комплексно.
Так как это генетическая патология, то терапия будет направлена на устранение появившихся симптомов:
- Назначается гормональное лечение. Это поможет в работе эндокринной системы. Необходимо внимательно следить за деформацией костей черепа, чтобы вовремя предотвратить развитие патологии.
- Оперативное вмешательство. Такой способ применяют, когда деформация лицевых костей может привести к нарушению слуха и зрения.
- Назначают обезболивающие препараты с содержанием бисфосфонатов.
Для каждого больного, страдающего от синдрома Олбрайта, способ терапии подбирается в индивидуальном порядке. Пациент должен строго соблюдать все рекомендации и назначения врача.
Проводится периодический контроль уровня кальция в организме пациента. На начальном этапе лечения такое исследование проводится один раз в неделю. Постепенно частота замеров снижается до одного раза в месяц. Она будет проводиться до конца назначенного лечения.
Если диагностирован псевдогипопаратиреоз, тогда больному необходимо придерживаться строгой диеты. Она заключается в полном исключении из меню продуктов, в которых присутствует фосфор.
Олбрайта синдром (F. Albright) - это заболевание, характеризующееся триадой признаков: фиброзная остеодисплазия, очаги кожной гиперпигментации и преждевременное половое созревание. Значительно чаще встречается у женщин.
Этиология синдрома Олбрайта неизвестна; предполагают неврогенные влияния и эндокринные сдвиги, вызванные врожденными изменениями в гипоталамической области, с преждевременной продукцией гонадотропного гормона передней доли гипофиза на почве склероза и гиперостоза основания черепа» главным образом в области турецкого седла (рис.).
Рост и зрелость скелета (появление и слияние ядер окостенения) могут значительно опережать нормальные показатели. Вследствие этого носители синдрома Олбрайта в детстве могут быть выше, а после преждевременного слияния эпифизов - ниже здоровых сверстников. Опережению развития скелета сопутствует преждевременное половое созревание - иногда стертое, редко значительное, когда менструальная функция может наступить в первые годы, а спорадические меноррагии - даже в первые месяцы жизни. Преждевременная половая зрелость у мальчиков редка.
Очаги гиперпигментации кожи при синдроме Олбрайта наблюдаются на туловище, бедрах, реже на шее, голенях. Они имеют вид крупных полей или отдельных и сливающихся пятен диаметром от 2 до 20 см и более, неправильной формы с изрезанными краями или представлены скоплениями веснушек от бледно-желтого до коричневого цвета. Иногда эти очаги видны уже при рождении, но при малой величине и бледности могут быть просмотрены. По локализации кожные изменения не всегда совпадают с костными поражениями. Гиперпигментированные участки кожи не приподняты.
При синдроме Олбрайта встречаются и другие отклонения - гипертиреоз, диабет, пороки сосудов, хрящевая остеодисплазия и др. Биохимические показатели при синдроме Олбрайта нормальны; иногда повышена активность щелочной фосфатазы (до 20 ед. Боданского). Среди осложнений фиброзной дисплазии, специфических для синдрома Олбрайта, встречаются атрофия зрительного нерва и экзофтальм как следствие гиперостоза области турецкого седла и орбиты.
Прогноз при синдроме Олбрайта благоприятный, однако возможны патологические переломы и деформация костей и в редких случаях озлокачествления фиброзных фокусов.
Дифференциальная диагностика и лечение костного компонента синдрома Олбрайта - см. Остеодисплазия. Кожные и эндокринные проявления при синдроме Олбрайта необратимы.
Уплотнение костной структуры основания черепа, преимущественно в области турецкого седла.
Псевдогипопаратиреоз (синдром Олбрайта) - это заболевание, характеризующееся морфологическими изменениями, гипокальциемической тетанией и отсутствием реакции на введение паратгормона. При заболевании отмечается низкорослость, коренастое телосложение, круглое лицо, преждевременное закрытие эпифизарных ядер окостенения, ожирение, брахидактилия, иногда короткие плюсневые кости, дистрофические изменения зубов и ногтей. Иногда наблюдается отставание интеллекта, кальцификация мягких тканей. В отличие от истинного гипопаратиреоза гипокальциемия, гиперфосфатемия и гипофосфатурия не корригируются введением паратгормона.
Заболевают обычно люди молодого и зрелого возраста.
Олбрайт объяснял это состояние рефрактерностью эпителия канальцев почек к действию эндогенного и экзогенного паратгормона, что приводит к повышенной обратной реабсорбции фосфора почками с гиперфосфатемией, гипофосфатурией, гиперкальциемией и гиперкальцийурией. Он относил псевдогипопаратиреоз к другим аналогичным состояниям органной рефрактерности к определенным гормонам (см. Сибрайт-бантамский синдром).
При этом заболевании околощитовидная железа имеет нормальное строение.
Псевдопсевдогипопаратиреоз - описанное в 1952 г. Олбрайтом патологическое состояние, напоминающее псевдогипопаратиреоз с присущими ему морфологическими изменениями, но без соответствующих гуморальных изменений.
Нарушения в эндокринной системе
Чаще всего у девочек, имеющих синдром Олбрайта наблюдается преждевременное половое созревание, которое вызвано эстрогенами, выделяющимися в кровь от кисты яичников. Кисты могут увеличиваться, после уменьшаться в своих размерах в течение нескольких недель или дней. С помощью процедуры УЗИ есть возможность увидеть и измерить размеры новообразований. Кисты могут вырастать до вполне приличных размеров. Были случаи, когда она выросла до размеров мяча для гольфа, то есть больше 50 мм в диаметре.
Увеличение груди и менструальные кровотечения наблюдаются вместе с ростом кисты. Если у девочки началась менструация до 2 лет, то это является первым симптомом синдрома Олбрайта. Однако наличие нерегулярных менструаций и кист яичников могут наблюдаться как у подростков, так и у взрослых женщин. Это все не мешает иметь здоровых детей.
Лечение детей, имеющих преждевременное половое созревание, достаточно трудно и малоэффективно. Даже если кисту удалят хирургическим способом, она может снова возникнуть. При принятии гормона прогестерона может быть остановлена менструация, но быстрые темпы развития и роста костей не замедляются. Возможны негативные последствия для работы надпочечников. При лечении используются препараты для приема внутрь, блокирующие синтез эстрогена.
Функции щитовидной железы
50 процентов людей, имеющих синдром Олбрайта, страдают нарушениями функции щитовидной железы. Это так называемый зоб, узелки и кисты. Возможны в редких случаях тонкие структурные изменения. У этих пациентов уровень вырабатываемого гипофизом тиреотропного гормона низкий, а вот уровни гормонов щитовидной железы имеют нормальные показатели или немного повышены. Проводится лечение, с помощью которого уменьшается синтез гормонов щитовидной железы. Оно показано в тех случаях, когда уровень выделяемых гормонов достаточно высок.

Чрезмерная секреция гормона роста
При заболевании гипофиз начинает выделять большое количество гормона роста. У детей, которым был поставлен такой диагноз, как синдром Олбрайта, была обнаружена акромегалия. У юношей стали появляться грубые черты лица, быстро увеличились руки и ноги, они могли страдать артритом. Лечение детей с такими признаками сводится к хирургическому удалению области гипофиза и использованию синтезированных аналогов гормона соматостатина, подавляющих выработку гормона роста.
Прочие эндокринные нарушения
Достаточно редко имеется чрезмерная секреция и расширение надпочечников. Подобное нарушение может привести к ожирению туловища и лица, увеличению массы тела, прекращению роста и кожной хрупкости. Все эти симптомы назвали синдромом Кушинга. При таких изменениях удаляют пораженный надпочечник или принимают лекарства, которые уменьшают синтез кортизола.
Иногда у детей, которые имеют синдром Олбрайта, в крови наблюдается очень низкий уровень фосфора вследствие большой потери фосфатов с мочой. Это нарушение может быть причиной изменения костей, связанного с рахитом. В качестве лечения назначают пероральные фосфаты и витамин D в дополнение.
Нарушения, связанные с кожей
На коже с рождения или вскоре после него появляются пятна цвета «кофе с молоком». Они чаще всего возникают на крестце, туловище, конечностях, ягодицах, задней поверхности шеи, лбу, на волосистой части головы, затылке. Все они также являются признаком того, что у ребенка синдром Олбрайта. Фото этих пятен можно увидеть ниже.

Хотя при таком заболевании, как нейрофиброматоз, тоже есть пятна цвета «кофе с молоком». Однако для синдрома Олбрайта характерны более крупные пятна с неправильными очертаниями, их меньше по количеству. Они имеют диаметр от 1 до нескольких сантиметров, коричневый оттенок. Цвет у всех одинаковый, они овальные по своей форме, для них характерна гладкая поверхность. При гистологических исследованиях чаще всего выявляется, что эпидермис не изменен по своей структуре, но количество меланина в кератиноцитах немного увеличено.
Единичные пятна подобного типа могут встречаться и у вполне здоровых людей. Если они не беспокоят и не растут, то в лечении нет необходимости. Если же наблюдается интенсивный рост, имеются пятна неправильной формы, то рекомендуется исследовать их гистологически. А затем удалить хирургическим способом.

Заключение
Таким образом, можно сказать, что для синдрома Олбрайта характерно поражение костей или черепа, наличие на коже пигментных пятен, раннее половое созревание. Хотя бывают случаи, когда присутствуют только первые два симптома. Вообще основным признаком синдрома являются костные поражения (остеодисплазия). Однако при наступлении половой зрелости этот процесс приостанавливается. У взрослых людей изменения костей не прогрессируют. Вообще, при выявлении и правильном лечении прогноз терапии данного заболевания вполне благоприятный.