Симптомите, които се появяват, се определят изцяло от клетъчното местоположение на мутиралия ген. При някои пациенти диагнозата се потвърждава в ранна детска възраст, тъй като са налице очевидни аномалии в развитието на костите и повишена работаендокринни жлези (например, менструацията може да започне в ранна детска възраст - спорадична менорагия).
Левкоплакия – причини за възникване при деца и възрастни, диагностика, локализация и методи на лечение
Ентеровирусна инфекция - причинител и пътища на предаване, симптоми, диагностика, методи на лечение и профилактика
Синдром на Dressler - причини за възникване след инфаркт, симптоми, диагноза, методи на лечение и профилактика
Болестта на Олбрайт се характеризира с три основни признака, ако два от трите са налице, тази диагноза вече може да бъде поставена:
- Преждевременно пубертет. Симптомът може да се прояви в пълна или непълна степен. Например, менструацията започва много рано, те са обилни, болезнени, но ако симптомът е непълен, може да ги няма изобщо. По време на кърмаческа възраст се появяват вторични полови белези, като подуване и развитие на млечните жлези, а често се появяват и кисти на яйчниците. При момчетата растат пубисни косми и пенис.
- Фиброзна остеодисплазия. Растежът на скелета, неговата зрялост и осификация са значително по-напред стандартни показателиразвитие. Нарушаването на костната тъкан води до изкривяване на костите, тяхната деформация и асиметрия е характерна. Най-честите признаци са куцота и изкривяване на гръбначния стълб. Растежът се забавя, което се забелязва още в детство, тогава ниският ръст също започва да придружава наднорменото тегло. Много пациенти изпитват неравномерен растеж на черепа, външна деформация и изпъкнали очи. Във възрастта на пубертета промените в костите се забавят и броят на фрактурите намалява.
- Промени в кожата. Това се изразява в хиперпигментация. Кожата на тялото е покрита с големи жълто-кафяви петна с назъбени ръбове. Областите на пигментация могат да бъдат разположени от засегнатата страна в лумбалната област, на бедрото, горната част на гърба, задните части и гърдите. Детето има светлина кафяви петнаобикновено се появяват веднага след раждането.
Синдромът на Олбрайт се проявява с признаци на увреждане на костната тъкан, кожата и симптоми на ендокринни нарушения.
- Пациентите развиват фиброзна остеодисплазия, проявяваща се с различни деформации и изкривяване на скелета. Най-често се засягат тръбните кости на един крайник, което води до куцота и деформация на гръбначния стълб. Още в ранна възраст образуването на скелета е нарушено. При пациентите се наблюдава деформация на челюстта и черепа: лицевите кости се удебеляват, стават широки, груби и големи. Така се образуват външни деформации: асиметрично лице и едностранен екзофталм. Тогава се появяват симптоми на артрит. Фиброкистозна дисплазия е фокално заместване на кортикална кост от пролифериращи фибробласти. Образуването на кисти води до костна деформация и фрактури. Най-честата деформация на тазобедрената става е тип „овчарски мошеник“.
- По кожата на пациентите се появяват области на хиперпигментация, които представляват групи от петна с неправилна форма и неравни ръбове. Цветът на петната често варира от жълтеникав до тъмнокафяв. Лезиите са локализирани по краката, бедрата и торса. Петната са достатъчно големи размерии гладка повърхност. Те присъстват върху кожата от раждането или се разпространяват, докато детето расте. Епидермисът не се променя. Техен интензивен растежизисква хистологично изследване и хирургическа намеса.
- Ендокринните нарушения са причинени от дисфункция на структурите на хипоталамо-хипофизната област и ендокринните жлези, разположени в периферията. Пубертетът започва на шест месеца и завършва на седем години. При пациентите започва менструация, развиват се млечни жлези и се развиват вторични полови белези - растеж на гърдите и поява на пубисно окосмяване. При ултразвуково изследване на яйчниците в тях се откриват големи фоликуларни кисти. В същото време самите яйчници остават нормални по размер.
- Пациентите изпитват тонични конвулсии, които възникват самостоятелно или под влияние на ендогенни и екзогенни стимули - стрес, хипо- или хипертермия, прекомерно пренапрежение.
- Калцификатите в кожата и меките тъкани често се възпаляват и образуват язви, причинявайки дискомфорт на пациентите.
- Възникват остеохондроми, хондроматоза и субпериостална резорбция на малките кости на пръстите.
- Типични външни признаци са нисък ръст, лунообразно лице, наднормено телесно тегло, къси пръсти.
- Излишъкът от фосфор пречи на усвояването на магнезия, причинявайки мигрена, аритмии и болки в гърба.
- Конфигурацията и подвижността на ставите се нарушават, образуват се фалшиви стави.
- Други признаци на синдрома включват: повръщане, хематурия, помътняване на лещата, частично отсъствие на зъбния емайл.
- Неврологичните разстройства са причинени от атрофия оптичен нерв, загуба на слуха и зрението.
- Забавено психомоторно развитие.
Има атипична или асимптоматична форма на псевдохипопаратироидизъм, при която количеството на калция и фосфора в кръвта остава нормално, няма гърчове или остеомалация.
Причини за синдрома на Олбрайт
Синдромът на Олбрайт е наследствена патология с неясен произход. Нарича се още псевдохипопаратироидизъм. Характеризира се с увреждане на костната тъкан. Болестта се проявява предимно в женската половина от населението, въпреки че се среща и при момчетата. Почти всички пациенти имат отклонения в умственото и физическото развитие.
Човешките кости започват да се развалят под въздействието метаболитни процесифосфор и калций. Започва отделянето на паратироиден хормон от паращитовидната жлеза.
Основната причина за такава патология като синдрома на Forbes-Albright е наследствената устойчивост на телесните тъкани към влиянието на паратиреоидния хормон. Ако човек не е болен, тогава хормонът действа чрез определено вещество. Когато заболяването се прояви, съставът на това вещество се променя на генетично ниво. Така периферните тъкани стават нечувствителни към паратироидния хормон.
Основни прояви
Всички прояви на това заболяване могат да бъдат разделени на няколко вида. Например ендокринните прояви включват:
- Нисък ръст.
- затлъстяване.
- Повишена кръвна захар.
- Лунообразно лице.
- Олигоменорея.
В някои случаи псевдохипопаратеозата се комбинира с акромегалия, синдром на Кушинг и гинекомастит.
Костно-ставните прояви на патология се изразяват в:
- Скъсяване на размера на пръстите.
- екзостози.
- Дисхондроплазия.
- Забавено никнене на зъби.
- Хипоплазия на зъбния емайл.
- Подкожни осификации.
Симптомите на псевдохипопаратироидизма се изразяват и в нарушения на нервната система. В същото време децата могат да получат конвулсии от раждането, както и тежка умствена изостаналост.
Други симптоми често включват уролитиаза, кръв в урината, повръщане и катаракта.
Ендокринните нарушения причиняват дисфункция на хипофизната жлеза и периферните ендокринни жлези.

Най-честите нарушения на половото развитие се характеризират с ранно съзряване. Преждевременното полово развитие се развива от шест месеца до седем години, в зависимост от индивидуалните характеристики на тялото. Тази патология се проявява в пълна форма и непълна форма.
Пълният външен вид се характеризира с наличието на два признака: ранното начало на менструацията и развитието на млечните жлези. При ненавършена бременност е характерна проявата на вторични полови белези без менструация. По този начин може да се появи ускорен растеж на височина, но в репродуктивна възраст такива момичета не се различават от жените с нормално развитие.
Ендокринните нарушения причиняват дисфункция на хипофизната жлеза и периферните ендокринни жлези
Ако пълната форма възниква с ускорено развитие, тогава непълната форма на развитие се удължава няколко пъти.
Синдромът на Олбрайт се различава от истинското твърде ранно развитие по това, че при хормонални дисфункции не се случва полов акт. растеж на косата, има леко ускоряване на скелетната диференциация.
В яйчниците се образуват големи фоликуларни кисти, но самите яйчници не се увеличават. Гонадотропните хормони обикновено остават непроменени.
Има и редица ендокринни патологии, включително хиперпаратироидизъм, хипертиреоидизъм, акромегалия, синдром на Иценко-Кушинг, рахит.
Възможна е фиброзна остеодисплазия, която се проявява с промени в осификацията, различни деформации и кривина на скелета. Не е необичайно синдромът на Олбрайт да включва увреждане на дългите кости от едната страна на скелета, което води до куцота и деформация. гръбначен стълб– това състояние може да се види на много снимки в интернет.
Темповете на растеж и формирането на скелета в много случаи се различават още в ранна възраст. По-голям процент от пациентите имат деформация на челюстните кости и черепа, което води до необичаен растеж на лицето, което поради хормонални промениводи до акромегалия (заболяването се характеризира с удебеляване на ръцете и лицевата част на черепа, а също така стават твърде широки) и други патологии, проявяващи се във външни деформации.
По този начин, по време на изследване, при деца с диагноза акромегалия, появата на твърде груби черти на лицето, по-ниски и Горни крайницибяха значително увеличени по размер, в допълнение към текущото заболяване се появиха симптоми на артрит.
От страна на кожата се забелязва хиперпигментация, която се проявява под формата на струпване на петна или плоски неправилни форми с неравни ръбове. Цветът на петната може да бъде жълтеникав или кафяв. Локализацията на хиперпигментацията е предимно по краката, бедрата или торса.
Фактори, допринасящи за развитието на синдрома:
- вродени аномалии на хипоталамуса,
- психогенни фактори - стрес, конфликти, неврози,
- ендокринопатии,
- хиперостоза на основата на черепа,
- преждевременна секреция на гонадотропини.
Синдромът на Олбрайт е резултат от нарушена функция на паращитовидните жлези. Основната им функция е регулирането на фосфорно-калциевия метаболизъм. При тази патология жлезите реагират слабо на вибрациите на тези елементи или изобщо не ги възприемат.
Генетично обусловена устойчивост структурни елементибъбреците и скелета към паратироидния хормон се причинява от увреждане на циторецепторите или дисфункция на аденилат циклазата. Мутацията на специфични гени води до потискане на производството на сАМР, който участва в метаболизма, който се осъществява под въздействието на паратироидния хормон.
Причините за дефицит на cAMP са различни:
- увреждане на нуклеотид-свързващите протеини на клетъчните мембрани,
- дефект на епителните циторецептори,
- разрушаване на аденилат циклазата.
Тези патологични промени правят периферните тъкани незасегнати от паратироидния хормон. Целевите органи губят чувствителност към паратироидния хормон, което води до повишаване на нивата на фосфор в кръвта и намаляване на концентрациите на калций.
Костна порьозност и образуване на кисти - характерни промениоткрити при този синдром. Калцият се измива от костите и се отлага под формата на калцификати в подкожната мастна тъкан, мускулите, вътрешните органи и ендотела на артериалните съдове. С време костензаменени с фиброзни. Скелетните аномалии нарушават подвижността на пациентите и причиняват силна болка.
Намаляването на витамин D в кръвта води до нарушаване на абсорбцията на калций в червата. Хипокалциемията стимулира още по-голямо освобождаване на паратиреоиден хормон в кръвта и повишено измиване на калций от костите, което води до развитие на тежка остеомалация. Костните нарушения при това заболяване се считат за най-тежките.
По кожата на пациентите се появяват пигментни петна кафявос назъбени ръбове. Ендокринните нарушения при това заболяване се изразяват в ранен пубертет и появата на вторични полови белези.
Ужасната диагноза, наречена синдром на Олбрайт, има неясна етиология.
Патологията с генетичен произход се проявява чрез повишена пигментация на кожата и промени хормонална системас ранно сексуално развитие. Рядко заболяване засяга цялото човешко тяло.
Много пациенти страдат не само от физическа изостаналост, пациентите с болестта на Олбрайт също се характеризират с умствени увреждания.
Псевдохипопаратироидизмът е и името на патологията, кръстена на американския учен Фулър Олбрайт, който открива това заболяване.
Честотата на заболяването се среща в световен мащаб като един случай на сто хиляди или един милион души.
Диагнозата е трудна и болестта на Олбрайт може да бъде погрешно диагностицирана, което затруднява точната оценка на нейното разпространение.
Синдромът на McCune-Albright (друг синоним на патология) се развива поради генетични мутации, които започват да се появяват в тялото случайно, без основателни причини.
Паращитовидната жлеза на тялото, която регулира нивата на калций и фосфор, секретира паратиреоиден хормон. При човек с този синдром тъканната резистентност към този хормон е нарушена.
В резултат на това човешката скелетна система започва да се влошава под влияние на промените в метаболизма на калция и фосфора.
Синдромът се характеризира с появата на специфични петна по тялото, фиброзна дисплазия и различни ендокринопатии. Сред последните най-разпространено е преждевременното полово развитие.
Някои генетици твърдят, че преобладаващата честота на пациенти сред момичетата, тъй като характерен симптомранен пубертет при момичетата се наблюдава почти 10 пъти по-често. Други прояви на синдрома се срещат почти еднакво и при двата пола.
Етиологията на синдрома е неизвестна.
Предполага се, че неврогенните влияния и ендокринните мутации са причинени от вродени аномалии в областта на хипоталамуса по време на ембрионалното развитие и ранното производство на гонадотропен хормон.
Учените смятат, че развитието на синдрома на McCune-Albright се дължи на липсата на реакция на бъбречните и костните клетки към действието на паратироидните хормони.
Нивата на калций и фосфор в кръвоносна системасе регулира от паращитовидните жлези, но при това патологично състояние техният интактен (безразличен) отговор на колебанията в тези елементи води до появата на хиперпаратироидизъм (прекомерно производство на паратироиден хормон) и промени в структурата на костите.
При синдрома на McCune-Albright неуспехът е причинен от генна мутация.
Прекъсване може да възникне навсякъде в метаболитната верига: при някои пациенти рецепторът е увреден, при някои пациенти протеините на клетъчната мембрана са претърпели промени, при други има недостатъчно производство на cAMP.
Генетичното заболяване, свързващо тези случаи, не е наследено: мутацията възниква след оплождането.
Симптоми
Симптомите, които се появяват, се определят изцяло от клетъчното местоположение на мутиралия ген.
При някои пациенти диагнозата се потвърждава в ранна детска възраст, тъй като има очевидни аномалии в развитието на костите и се отбелязва повишена активност на ендокринните жлези (например менструацията може да започне в ранна детска възраст - спорадична менорагия).
При други пациенти симптомите се появяват по-късно. Основен патологични признациса съпроводени с общо изоставане в развитието на човека – интелектуално и физическо.
Диагностика
Болестта на Олбрайт е трудна за диагностициране при кърмаче, а на възраст 6-10 години симптомите стават по-изразени. Има множество отклонения в развитието на костната система, калцият в кръвта е намален, фосфорът и паратиреоидният хормон са повишени.
Симптоми
Има три основни признака на заболяването. Ако са налице дори две от три, може да се постави такава диагноза.
- Много ранен пубертет в резултат на силно повишени хормонални нива. Момичетата могат да започнат менструация през първите месеци след раждането. Момчетата със синдром на Олбрайт много рядко изпитват ранен пубертет. Промени в хормоналните нива възникват поради неправилно функциониране на надбъбречните жлези, хипофизата и щитовидната жлеза. Този симптом може да се появи в пълна или непълна форма. При пълна форма менструацията започва много рано и обикновено е обилна и болезнена. IN младенческа възрастПри този синдром се появяват и вторични полови белези, например подуване на млечните жлези. Ако изявата е непълна, менструацията не настъпва. Много често със синдрома на Олбрайт момичетата развиват кисти на яйчниците. При момчетата по време на ранния пубертет се наблюдава уголемяване на пениса и окосмяване на пубиса.
- Фиброзна остеодисплазия. Това заболяване показва нарушение на костната тъкан, което води до изкривяване и деформация на костите. Характеризира се с асиметрия. Най-честите признаци са куцота и изкривяване на гръбначния стълб. Най-често от промени са засегнати дългите кости. Растежът на скелета се забавя, това се забелязва дори при деца. Много жертви на синдрома на McCune изпитват неравномерен растеж на костите на черепа, което води до външна деформация. Когато лицето се промени, може да се появят изпъкнали очи. След пубертета промените в костите се забавят и честотата на фрактурите намалява.
- Промени в кожата. Проявява се в повишена пигментация. Кожата на торса и бедрата се покрива с жълто-кафяви петна с назъбени ръбове. Пигментация може да се види в долната част на гърба, горната част на гърба, задните части и гърдите. Петната се появяват веднага след раждането на бебето.
Синдромът на Олбрайт е наследствено заболяване, което включва комплекс от патологии на ендокринната система, кожата и опорно-двигателния апарат. Понякога това заболяване се нарича псевдохипопаратироидизъм, наследствена остеодистрофия или „синдром на яванското пиле“.
През 1942 г. тази патология е документирана за първи път от американския лекар Фулър Олбрайт, който изучава ендокринната система, по-специално калциевия метаболизъм.
Синдромът на Олбрайт е много рядък, приблизителната честота на заболяването е 7,9 на 1 милион души, като патологията е по-често срещана при жените.
Всяка наследствена патология се причинява от отрицателни мутации в гените, което причинява смущения в развитието на всяка телесна система, свързана с функционирането на тези гени.
Болестта на Олбрайт се характеризира с мутации на гена GNAS1, който кодира специален протеин Gs - алфа. Този протеин е пряко свързан с рецепторите на паратироидния хормон (PTH).
Унаследяването на остеодистрофията на Олбрайт е свързано с Х-хромозомата, което се доказва от липсата на наследяване от баща на син и високата честота на заболяването при жените.
Намаляването на активността на GS-алфа протеина води до факта, че телесните тъкани стават нечувствителни към паратироидния хормон. В допълнение, синтезът на cAMP, молекула, която действа като посредник за разпространението на сигнали на определени хормони, е нарушен в клетките.
Симптоми
Брахидактилия при синдрома на Олбрайт Болестта се проявява като нисък ръст лице с форма на луна
В зависимост от причината за патологията се разграничават няколко вида на заболяването.
Псевдохипопаратироидизмът, който се характеризира с патология на клетъчните рецептори, които свързват паратироидния хормон, се класифицира като заболяване тип 1а. Друг тип заболяване, 1b, се характеризира с разрушаване на протеини, които свързват рецепторите и ензимите помежду си.
Псевдохипопаратиреоидизъм тип 2 се характеризира с остър дефицит на сАМР.
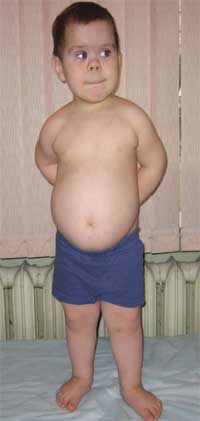
Независимо от вида на заболяването, болестта на Олбрайт се характеризира с определена общи симптоми. Поради нарушаване на развитието на ендокринната система, пациентите могат да получат увеличение на паращитовидните жлези. Тялото може да се развие непропорционално поради скъсяване долните крайници, което води до забележим нанизъм (вижте снимката по-горе).
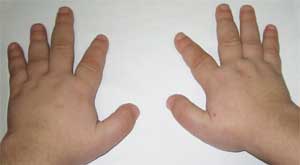
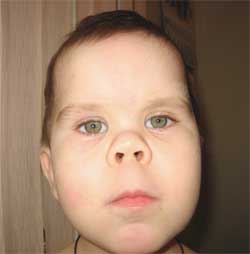
Пръстите също могат да се скъсят, което се нарича брахидактилия. Лунообразна форма на лицето. Може да се появи затлъстяване и остеопороза. Поради нарушеното развитие на опорно-двигателния апарат може бързо да се развие деформация на гръбначния стълб, появата на фалшиви стави и липса на подвижност в онези области, където би трябвало да е естествено анатомично.
Често пациентите с болестта на Олбрайт развиват синдром на Иценко-Кушинг
Поради неправилното развитие на костната тъкан възникват проблеми със зъбите поради бързо увреждане на емайла. Поради хипокалциемията съществува висок риск от развитие на катаракта.
Синдромът на Олбрайт също се характеризира с калцификация на меките тъкани поради калций, който е бил освободен от костите.
Калциевите отлагания могат да се образуват в бъбреците, различни мускули, по стените на кръвоносните съдове и в миокарда, което може да доведе до сърдечни усложнения.
Като цяло, калцификатите, дори когато са разположени в подкожната тъкан, могат да засегнат тъканите около тях, което води до образуването на малки, но неприятни язви.
В допълнение към горните промени се появяват петна по кожата на пациента различни размерицвят кафе (вижте снимката по-долу).
Нарушение на пигментацията на кожата (по тялото се появяват кафяви петна, подобни на бенка)
Отделна точка в списъка на неврологичните разстройства са тоничните гърчове. Те могат да възникнат както сами, така и поради действието на всякакви дразнители, например стрес, изтощителна физическа работа, внезапна промянатемператури и др.
Диагностика
Тъй като псевдохипопаратироидизмът е наследствено заболяване, ако има хора с подобна патология, особено близки роднини, е необходима консултация с генетик, за да се оцени рискът от раждане на болно дете.

Доста трудно е веднага да се диагностицира синдром на Олбрайт при новородено, но в детството (5-10 години) това вече става възможно, тъй като се появяват всички симптоми, характерни за заболяването.
Необходимо е също така да се проведат редица изследвания, по-специално кръвен тест за определяне на нивото на калций, фосфор и паратиреоиден хормон. Тестът за урина може да определи количеството фосфор и сАМР.
Има редица специални диагностични тестове, насочени към изследване на чувствителността на тъканите към паратироидния хормон.
Рентгеновата диагностика може да покаже промени в скелета и комбинацията от резултати от тестове, физически и психически симптоми ще покаже какъв вид заболяване е и дали може да бъде синдром на Олбрайт, тъй като подобни симптоми могат да се наблюдават при такива наследствени заболявания като Шерешевски- Синдром на Търнър.
Много е важно да се ранна диагностикапсевдохипопаратироидизъм, което води до бързо започване на лечението. Колкото по-бързо започне необходимото лечение, толкова по-малък е рискът от аномалии в умственото развитие. Обикновено пациентите със синдром на Олбрайт се наблюдават от няколко специалисти:
- гинеколог или уролог;
- рентгенолог;
- ендокринолог;
- ортопед;
- диетолог и др.
Това е свързано със сложни симптоми на патологията.
Почти винаги на пациентите се предписват различни калциеви добавки, тъй като, за разлика от пациентите с истински хипопаратироидизъм, инжекциите на паратиреоиден хормон не водят до желан ефект.
Много е важно да изберете правилната доза, за да поддържате нормална хомеостаза.
За по-пълно усвояване на калций, заедно с лекарства, базирани на него, те се предписват витаминни комплекси, особено тези, съдържащи витамин D, като:
- Оксидевит;
- калцитриол;
- дихидротахистерол.
В допълнение към приема на лекарства, пациентите трябва да спазват строга диета, също базирана на храни, богати на калций.
Не трябва да се допуска обратен ефект– прекомерни нива на калций над нормата, тъй като може да се появи хиперкалциемия, придружена от загуба на сила, главоболие и стомашно-чревни разстройства.
IN в такъв случайНеобходимо е напълно да се изоставят калциевите добавки и преди всичко да се излекува хиперкалциемията.

Ако заболяването е придружено съществени нарушенияендокринната система, тогава може да се наложи да хормонална терапия. Тамоксифен, лекарство, което блокира естрогенните рецептори, често се използва за хормонална терапия.
Нивата на калций се проверяват всяка седмица, за да се види дали курсът на лечение трябва да се коригира или не. Впоследствие нивата на калций се проверяват веднъж месечно и след това веднъж на тримесечие, ако се постигнат благоприятни резултати.
При конвулсии се прилага интравенозно 10% разтвор на калциев хлорид или калциев глюконат. Много е важно разтворът да се прилага бавно, тъй като съществува риск от интоксикация. Препоръчва се венозните инжекции да се повтарят 2-3 пъти на ден.
Ако се наблюдават специфични и специфични нарушения, те се лекуват от подходящи специалисти, например катаракта - офталмолог, а зъбните заболявания - зъболекар.
При възрастни патологичните промени в костната тъкан прогресират.
хирургия
Хирургията за синдрома на Олбрайт се извършва, когато анормалното развитие на костите може да увреди нерва.
Диагностика
Етиопатогенетични фактори
Псевдохипопаратиреоидизмът се основава на наследствената устойчивост на телесните тъкани към влиянието на паратироидния хормон. При нормални условия последният действа чрез специален посредник - цикличен аденозин монофосфат (цАМФ). Той е този, който инструктира клетката да се подчини на регулаторния сигнал на паратироидния хормон. При болестта на Олбрайт структурата на медиатора се променя на генетично ниво.
Болестта на Олбрайт се предава на следващите поколения по интересен модел, който се нарича Х-свързано доминантно унаследяване: патологията преминава от майката към дъщерите и синовете в 50% от случаите. Със същата вероятност само дъщерите наследяват дефектния ген от баща си. Има фамилни случаи на заболяването.
Генът на синдрома на Олбрайт се намира на половата хромозома
Без регулаторното действие на паратироидния хормон метаболизмът на калция се измества значително към използване в костната тъкан. В този случай има негов дефицит в кръвта, което води до следните явления:
- мускулни крампи;
- нарушаване на образуването на електрически импулси в миокарда и промени в сърдечния ритъм;
- повишено производство на паратироиден хормон и уголемяване на паращитовидните жлези.
Излишъкът от паратироиден хормон, от своя страна, води до повишаване на нивата на фосфор в кръвта.
Щитовидната и паращитовидните жлези са основните регулатори на метаболизма на калция и фосфора в организма.
Това генетична патологияхарактеризиращ се с автозомно-доминантен начин на унаследяване.
Основната причина е вроден дефект, който прави периферните тъкани нечувствителни към паратироидния хормон.
В зависимост от това как и къде е нарушена метаболитната верига, се разграничават два вида синдром.
Лечение на заболяването
Типичните видими признаци включват:
- нисък ръст;
- наднормено тегло;
- високи нива на глюкоза;
- лице с форма на луна;
- нарушения на пубертета.
Могат да се появят и следните симптоми:
- акромегалия;
- автоимунни заболявания;
- гинекомастия;
- Болест на Иценко-Кушинг и др.
Пациентите често се оплакват от кръв в урината, постоянно повръщане, промени в зъбите и гърчове. При преглед се вижда подкожна осификация, може да се появят калцификации.
Пациентите често имат скъсени пръсти, деформирани епифизи на дълги кости и може да има признаци на лещовидна катаракта.
Почти винаги такива пациенти изостават в умственото развитие.
Псевдохипопаратироидизмът се лекува комплексно.
Тъй като това е генетична патология, терапията ще бъде насочена към премахване на симптомите, които се появяват:
- Назначава се хормонално лечение. Това ще помогне за функционирането на ендокринната система. Необходимо е внимателно да се следи деформацията на костите на черепа, за да се предотврати развитието на патология навреме.
- хирургия. Този метод се използва, когато деформацията на лицевите кости може да доведе до увреждане на слуха и зрението.
- Предписват се болкоуспокояващи, съдържащи бифосфонати.
За всеки пациент, страдащ от синдрома на Олбрайт, методът на лечение се избира индивидуално. Пациентът трябва стриктно да спазва всички препоръки и предписания на лекаря.
Периодично се проследяват нивата на калций в тялото на пациента. В началния етап на лечението такова изследване се провежда веднъж седмично. Постепенно честотата на измерванията се намалява до веднъж месечно. Ще се провежда до края на назначеното лечение.
Ако се диагностицира псевдохипопаратиреоидизъм, пациентът трябва да се придържа към строга диета. Състои се в пълно премахване от менюто на храни, които съдържат фосфор.
Наследственото заболяване най-често засяга цялото тяло, което води до множество промени. Признаците на патология в този случай са изключително сходни с тези на хипопаратироидизма - дефицит на производство на паратироиден хормон в паращитовидните жлези.
Синдром на Олбрайт - генетично заболяванес неизвестна етиология, засягащи цялото човешко тяло. Патологията се среща сравнително рядко и се проявява чрез асиметрична хиперпигментация на кожата, хормонален дисбаланс и полиостозна фиброзна костна дисплазия. Това заболяване е рядка форма на гонадотропин-зависим ранен пубертет.
Синдромът на Олбрайт се развива предимно при жени справедлива половиначовечеството. Причинява се от спонтанна генна мутация по време на ембриогенезата и не се предава по наследство. Диагнозата се поставя при деца на възраст 5-10 години въз основа на признаци, характерни за това заболяване.
Клиничната картина на синдрома е описана за първи път през 1907 г. от учения Олбрайт, на когото той получава името си. Ученият наблюдавал ниски и набити момичета с кръгло лице и къс врат. Установено е, че те имат мускулни спазми, скелетни деформации, частична липса на зъбен емайл, умствена изостаналост и признаци на затлъстяване.
Ендокринопатията се проявява с ранен пубертет: поява на менструация, активен растеж млечни жлези, появата на пубисно окосмяване. Олбрайт обяснява произхода на патологията с резистентността на бъбречния епител към паратироидния хормон, което води до ускорена реабсорбция на фосфор и развитие на хиперфосфатемия.
В същото време паращитовидните жлези при пациентите имат нормална структура и няма хуморални промени.
IN официална медицинаСиндромът се нарича псевдохипопаратироидизъм. Нарушенията на фосфорно-калциевия метаболизъм водят до патология на скелетната система. Такива нарушения могат лесно да се обяснят с резистентност на тъканите към ендогенния паратироиден хормон. Пациентите имат изоставане в психофизическото развитие. В този случай няма реакция към въвеждането на екзогенен паратироиден хормон.
Пациентите със синдром на Олбрайт се нуждаят от консултация със специалисти в областта на гинекологията, ендокринологията, радиологична диагностика, офталмология, травматология и ортопедия.
Това заболяване се среща предимно при момичета.
Нарушенията се появяват в резултат на генна мутация в ембрионалния стадий на развитие и не могат да се предават по наследство. Чертите, които се появяват, идват изцяло от клетъчното местоположение, което съдържа мутиралия ген.
Диагностика
Липсата на повишена бъбречна резистентност към паратироидния хормон е достатъчна основа за диагностициране на болестта на Олбрайт.
Биохимичният кръвен тест, който разкрива хипофосфатурия (недостатъчно отделяне на фосфор в урината), се допълва от рентгеново изследване за проследяване на промените в костите и меките тъкани.
Жените трябва да преминат преглед от гинеколог, ехографияанализ на таза и половия хроматин. Фокусите на петна са видими при раждането, но поради малкия си размер и бледостта си често не се забелязват.
Предотвратяване
Превенцията е както следва:
- хората с тази патология трябва редовно да правят физически упражненияза укрепване на мускулите;
- ако една жена иска да забременее, тогава трябва да се консултира с гинеколог и генетик.
Тази патология се диагностицира при един човек на един милион души на планетата. За съжаление днес няма специфична терапия. Има само симптоматично лечение. Но ако пациентът е под наблюдението на лекар, има шанс за благоприятен изход.
Симптоми
- Поради високите хормонални нива настъпва ранен пубертет. Момичетата могат да започнат менструация през първите десет месеца от живота си. Но този симптом не се отнася за момчетата. Те започват пубертета както обикновено. Хормоналните нива се повишават, когато надбъбречните жлези, щитовидната жлеза и хипофизата не функционират правилно. Този симптом може да се развие в две форми - непълна и пълна. Ако патология пълна форма, тогава вашият цикъл започва твърде рано. В същото време те са болезнени и изобилни. Появяват се и вторични полови белези, например уголемяване на млечните жлези. Ако формата на патологията е непълна, тогава менструацията не започва. Болестта на Олбрайт при момичетата често е придружена от развитие на кисти на яйчниците. Ранният пубертет при момчетата се характеризира с уголемяване на гениталния орган и поява на пубисно окосмяване.
- Фиброзна остеодисплазия. Това е патология, при която костната тъкан се деформира, което води до огъване на костите на скелета. Проявява се под формата на асиметрия. Визуалните симптоми са гръбначно изкривяване и куцота. Тази патология засяга предимно тръбните кости. Човешкият растеж се забавя. Това може да се забележи дори при дете. Костите на черепа се развиват неравномерно и се появяват изпъкнали очи. След края на пубертета промените в костите спират.
- Промяна в цвета на кожата. Започва да се появява силна пигментация. По бедрата и самото тяло се появяват жълто-кафяви петна с размити ръбове. Петната са ясно видими в долната част на гърба, горната част на гърба, гърдите и задните части. При синдрома на Олбрайт петна се появяват по тялото на детето веднага след раждането.
Заболяването псевдохипопаратироидизъм се проявява със специфични симптоми.
Вроденото изкълчване на тазобедрената става е една от най-честите аномалии в развитието. Недоразвитие или дисплазия тазобедрена ставаима както едностранен, така и двустранен характер. Причините за развитието на патологията не са напълно изяснени, но клиницистите познават широк спектър от предразполагащи фактори, които могат да действат като провокатор на заболяването, вариращи от генетично предразположение до неадекватна бременност.
Остеохондропатия (припокриващи се симптоми: 2 от 11)
Остеохондропатия е сборно понятие, което включва заболявания, които засягат опорно-двигателния апарат, придружени от деформация и некроза на засегнатия сегмент. Трябва да се отбележи, че такива патологии са най-чести при деца и юноши.
Неврофиброма (припокриващи се симптоми: 2 от 11)
Неврофибромата е a доброкачествен тумор, образуван от нервна тъкан както в централната, така и в периферната нервна система. В по-голямата част от ситуациите се диагностицира при юноши и хора на средна възраст.
Болест на Шойерман-Мау (припокриващи се симптоми: 2 от 11)
Болестта на Шойерман-Мау (син. кифоза на Шойерман, дорзална (дорзална) ювенилна кифоза) е прогресираща деформация на гръбначния стълб, която се развива по време на активен растежтяло. Без навременно лечение може да доведе до сериозни последствия.
Меланоза (припокриващи се симптоми: 2 от 11)
Меланозата е голямо количество меланинов пигмент, натрупан в тялото. В определени области кожатапоявява се силна пигментация. Най-често патологията се диагностицира при жени в детеродна възраст. Проблемът причинява дискомфорт, тъй като се вижда визуално и лечението е трудно. Всяка започната терапия ще донесе само леко облекчение, а промените в начина на живот често се препоръчват като превантивни мерки.
Източник
Диагностика

Синдромът на Олбрайт се отличава не само с резистентността на бъбреците и скелета към паратироидния хормон, но и на други органи към съответните хормони - яйчниците към гонадотропните, щитовидната жлеза към тиреостимулиращите, надбъбречните жлези към антидиуретичните. Синдромът на Олбрайт се придружава от следните ендокринопатии: хипертиреоидизъм, акромегалия, синдром на Иценко-Кушинг.
- Дисфункцията на щитовидната жлеза се проявява с така наречената гуша, възли и кисти на органа.
- Прекомерното производство на хормон на растежа от хипофизната жлеза води до развитие на акромегалия. Чертите на лицето стават груби, ръцете и краката стават дълги, появява се артрит.
- Прекомерната секреция на надбъбречни хормони причинява затлъстяване, забавяне на растежа и крехкост на кожата - типични признациСиндром на Кушинг.
Наблюдава се и повишена честота на автоимунни заболявания, захарен диабет, съдови дефекти и хрущялна остеодисплазия.
Усложненията на синдрома на Олбрайт включват: оптична атрофия, екзофталмос, патологични фрактури, костна деформация, злокачествено заболяване на фиброзни огнища, кръвоизливи в различни части на тялото, кървене, смущения в червата, черния дроб, бъбреците и храносмилателната система.
Прогнозата на терапията с навременно разпознаване на болестта на Олбрайт е относително благоприятна, но е невъзможно напълно да се излекува наследственият ензимен дефект. Нормализирането на съдържанието на калций и фосфор в организма ще позволи на пациентите да се занимават с лека работа, с изключение на нервно напрежениеи работа с машини и механизми.

В случай на повтарящи се пристъпи на мускулни крампи, които трудно се коригират с лекарства, медицинската комисия взема решение за определяне на група инвалидност. При тежки случаи на псевдохипопаратироидизъм могат да се развият следните усложнения:

Обмяна на калций и фосфор в организма
Метаболизмът е най-важният аспект от живота на човешкото тяло. Всяка секунда в тъканите и органите едни химични елементи се превръщат в други. По правило тяхното съдържание е строго регламентирано. Това важи особено за електролитите – натрий, калий, калций и фосфор. Благодарение на техните специфични взаимоотношения е възможна работата на сърцето и скелетните мускули и предаването на нервен електрически сигнал.
Калцият се намира във всички органи и тъкани и играе няколко важни роли:

Фосфорът се намира в големи количества в костната тъкан и е от съществено значение за нормалната мозъчна функция. Калцият и фосфорът присъстват в кръвта в строго определено съотношение. Излишъкът от тези химични съединения се отделя от бъбреците с урината.
Паратироидният хормон е основният регулатор на нивата на калций и фосфор в кръвта

Благосъстоянието на тялото пряко зависи от постоянното количество калций и фосфор. Процесът на тяхната обмяна се контролира от щитовидната и паращитовидните жлези:
- Първият произвежда хормона калцитонин, благодарение на който калцият се прехвърля от кръвта в костната тъкан.
- Последните, използвайки паратиреоиден хормон, премахват липсата на калций в кръвта, като го измиват от костите.
В допълнение, обмяната на калций и фосфор се регулира от бъбреците, премахвайки излишъка от тези химични съединения в урината. Витамин D играе важна роля в това.
Синдромът на Олбрайт е наследствено заболяване, причинено от дефект в метаболизма на калция и фосфора в организма. Води до различни промени в костите, бъбреците, храносмилателния тракт и психични разстройства.
Наследствената остеодистрофия е разгледана за първи път от учения Фулър Олбрайт през 1942 г. Заболяването се среща няколко пъти по-често при жените. В медицинската литература са описани само около 300 случая на тази рядка патология.
Класификация
Псевдохипопаратироидизмът се разделя на три основни типа:

Диагностични методи
За разпознаване на болестта на Олбрайт е необходима съвместната работа на няколко специалисти: уролог, невролог, психиатър, ендокринолог, генетик. За установяване на правилна диагноза се използват следните методи:

Диференциална диагноза се провежда с следните заболявания:

Псевдохипопаратиреоидизмът изисква наблюдение през целия живот от ендокринолог. При повторен тежки атакиконвулсии изискват хоспитализация в специализирано болнично отделение.
Лечението с лекарства е основният компонент на терапията на псевдохипопаратироидизма. Основните цели са:
- нормализиране на нивата на калций и фосфор в кръвта;
- възстановяване на нормалната костна структура;
- предотвратяване на гърчове;
- предотвратяване образуването на камъни в бъбреците.
За нормализиране на нивата на калций се използват лекарствата Калциев хлорид и Калциев глюконат. Най-често тези лекарства се използват под формата на таблетки. Въпреки това, за да се премахнат мускулните крампи, се практикува интравенозно приложение. фармакологични средства.
Калциевият глюконат е лекарство, използвано за лечение на болестта на Олбрайт
Добавките с витамин D помагат за нормализиране на нивата на калций и фосфор в кръвта:
- Калций D3;
- Оксидевит;
- Калцитрин.
Калций D3 се предписва при болестта на Олбрайт
Прилагането на паратироиден хормон при псевдохипопаратироидизъм е неефективно, тъй като тъканите и органите са нечувствителни към неговото влияние.
Корекция на диетата
Важен компонент от лечението на псевдохипопаратироидизъм е корекцията на диетата. В същото време достатъчен калориен прием и дробни хранения. Количеството течност, което пиете на ден, трябва да бъде най-малко 1,5-2 литра.
- боб;
- зелен грах;
- леща за готвене;
- бадем;
- кресон;
- целина;
- карфиол;
- копър;
- босилек;
- мляко;
- кисело мляко;
- сметана.
Синдром на Олбрайт – рядко заболяване, което се предава по наследство и се проявява с увреждане на костната система. Причината за това е нарушение на метаболизма на калций и фосфор. Възниква поради резистентност на тъканите към паратироидния хормон, който се произвежда от паращитовидните жлези. Този синдром често е придружен от умствено и физическо изоставане в развитието.
Този симптом се нарича още псевдохипопаратироидизъм.
Това заболяване е описано за първи път от учения Олбрайт, в чиято чест е получило името си.
Видове
Има два вида псевдохипопаратироидизъм в зависимост от нивото на калций в кръвта:
- Първата клинична картина е много подобна на идиопатичния хипопаратиреоидизъм - ниски нива на калций и нечувствителност към паратироидния хормон.
- Вторият показва нормални нива на калций и затова се нарича псевдохипопаратироидизъм.
Лекарите отбелязват, че една форма на заболяването може да се трансформира в друга. Понякога това заболяване може да приеме различни форми сред членовете на семейството.
При мъжете тази патология причинява репродуктивна дисфункция, така че се предава по мъжка линия изключително рядко.
Причини и развитие
Тази генетична патология се характеризира с автозомно-доминантен начин на наследяване.
Основната причина е вроден дефект, който прави периферните тъкани нечувствителни към паратироидния хормон.
В зависимост от това как и къде е нарушена метаболитната верига, се разграничават два вида синдром.
Знаци
Типичните видими признаци включват:
- нисък ръст;
- наднормено тегло;
- високи нива на глюкоза;
- лице с форма на луна;
- нарушения на пубертета.
Могат да се появят и следните симптоми:
- акромегалия;
- автоимунни заболявания;
- гинекомастия;
- Болест на Иценко-Кушинг и др.
Пациентите често се оплакват от кръв в урината, постоянно повръщане, промени в зъбите и гърчове. При преглед се вижда подкожна тъкан, може да се появят калцификации.
Пациентите често имат скъсени пръсти, деформирани епифизи на дълги кости и може да има признаци на лещовидна катаракта.
Почти винаги такива пациенти изостават в умственото развитие.
Диагностика
Заболяването се диагностицира при деца на възраст от пет до десет години. Лекарят получава характерна клинична картина, идентифицира голям брой аномалии в развитието на скелета, наличието на:
- хипокалцемия;
- хиперфосфатемия;
- намалена екскреция на калций и фосфор в урината;
- повишени нива на паратироидния хормон;
- резистентност на бъбречните тубули и др.
Лечение
Лечението се състои от различни лекарствакалций, които са подбрани специално за поддържане на хомеостазата в нормално състояние.
Необходимо е също да се прилагат витамини D, калцитриол и оксидевит.
Индивидуално се разработва диета, която ограничава храните с високо съдържаниефосфор и допринася за образуването на нормални нива на калций в кръвта.
Ако има съпътстващи заболявания на ендокринната система, редица хормонални лекарства, като заместителна терапия.
Спазмите се елиминират с калциев глюконат или разтвор на калциев хлорид.
Ако подходите към лечението изчерпателно и правилно, то може да даде доста благоприятни резултати. Но всеки такъв човек, дори и да е излекуван и да води нормален живот, трябва да бъде прегледан от генетик, за да се установят рисковете, преди да има дете.
За първи път в руската литература този синдром е описан от В. Р. Брайцев през 1922 г., определяйки го като "фиброзни тумори" 15 години по-късно ендокринологът Фулър Олбрайт формулира характеристиките на това системно заболяване като „синдром, характеризиращ се с дисеминиран фиброзен остеит, пигментирани полета и ендокринни нарушения с преждевременен пубертет при момичетата“.
С други думи, синдромът на McCune-Albright-Braitsev е системно заболяване, характеризиращо се с преждевременно сексуално развитие и костна патология. Това заболяване се среща предимно при жените и е вродена наследствена патология. Доскоро се смяташе, че това е изключително женска болест, но по-късно са регистрирани случаи на заболявания и сред момчетата. Причината за синдрома се счита за мутация в специфичен ген.
Признаци на синдрома на McCune-Albright-Braitsev
- Тъмни петна.Всички пациенти имат специална пигментация с неправилна форма, цвят на кафе с мляко. Петната обикновено са разположени по гърба, гърдите, бедрата, задните части и долната част на гърба. Това е един вид маркер на заболяването, пигментацията присъства от раждането или се появява, когато детето расте.
- Костна патология.Дългите кости (като бедрената кост) обикновено са засегнати. Наличието на кухини в костите води до тяхната деформация, фрактури и огъване на крайниците. Типична деформация на тазобедрената става е типът „овчарски мошеник“. Възможно увреждане на костите на черепа. В този случай се появява асиметрия на лицето и едностранен екзофталм (изпъкнали очи). Уврежданията на костите и честотата на фрактурите намаляват след пубертета.
- Преждевременно сексуално развитие.Може да започне от 6-9 месечна възраст до 7 години. Най-често първите признаци на пубертета се появяват при дете на 3 години. Може да се прояви в пълна и непълна форма. Пълната форма при момичетата се характеризира с менархе (първа менструация) и уголемяване на млечните жлези. Възможно е продължително и обилно кървене, често се срещат и кисти на яйчниците. При непълна форма менархе не настъпва.
Преждевременният пубертет при момчетата е много по-рядко срещан и е съпроводен със симетрично уголемяване на тестисите, след това растеж на пениса и пубисно окосмяване, както при нормалното полово развитие.
- Ускоряване на костната възраст и ускорено затваряне на растежните пластини.
- Понякога се наблюдават неврологични и психични разстройства (забавяне умствено развитие, гърчове, оптична атрофия, загуба на слуха).
- В литературата има информация за др ендокринни нарушенияс това заболяване: акромегалия, хиперпаратиреоидизъм (повишена функция на паращитовидните жлези), хипертиреоидизъм (повишена функция на щитовидната жлеза) и наличие на възли в щитовидната жлеза, хиперкортизолизъм (излишък на глюкокортикоидни хормони на надбъбречните жлези), рахит, устойчив на витамин D.
Диагностика на синдрома на McCune-Albright-Braitsev
- Наличие на характерни черти.
- Генетична консултация.
- рентгеново изследване.
- Полови хормони и гонадотропини, хормони на щитовидната жлеза, пролактин, други тропични хормони на хипофизата (по показания).
- Ултразвук на тазови органи за момичета и скротум за момчета.
- Консултация с ендокринолог и детски гинеколог за момичета или детски урологза момчета.
- Изследване на други органи на ендокринната система (по показания).
Лечение на синдрома на McCune-Albright-Braitsev
Пациентите с тази патология се нуждаят от наблюдение от гинеколог-ендокринолог, ендокринолог, рентгенолог, офталмолог, ортопед-травматолог. Освен това е необходимо периодично да се определя нивото на тропните и гонадотропните хормони. Специфично лечение за този синдром не е разработено.Най-често лекарите избират тактика за наблюдение. За момичета терапията е показана в случай на продължително повишаване на нивата на естроген, придружено от чести и тежки кръвоизливи. По показания се извършва корекция на костни деформации. Ако се развият симптоми на увреждане на други ендокринни жлези, лечението се извършва от ендокринолози.
Catad_tema Педиатрия - статии
Псевдохипопаратироидизъм (наследствена остеодистрофия на Олбрайт): трудности при диференциално диагностично търсене. Клинично наблюдение
Е.В. Тозлиян, детски ендокринолог, генетик, д-р И.В. Шулякова, невролог, д-р,
отделна структурна единица „Изследователски клиничен институт по педиатрия“ на Държавната бюджетна образователна институция за висше професионално образование „Руски национален изследователски медицински университет на името на Н.И. Пирогов" на Министерството на здравеопазването на Руската федерация, Москва
Ключови думи:
деца, псевдохипопаратиреоидизъм, наследствена остеодистрофия на Олбрайт, затлъстяване, хипокалциемия, диагноза, резистентност към паратхормон.
Ключови думи:деца, псевдохипопаратиреоидизъм, наследствена остеодистрофия на Олбрайт, затлъстяване, хипокалцемия, диагностика, резистентност към паратиреоиден хормон.
Псевдохипопаратиреоидизъм (на гръцки pseudes - фалшив + хипопаратиреоидизъм; синоним: наследствена остеодистрофия на Олбрайт, синдром на яванското пиле) е рядко наследствено заболяване на скелетната система, симулиращо хипопаратиреоидизъм и характеризиращо се с нарушен метаболизъм на калций и фосфор; често придружени от забавено умствено и физическо развитие. Заболяването е описано за първи път от американския ендокринолог Олбрайт Ф. през 1942 г. Разпространението на заболяването е 7,9 на 1 милион души.
ГЕНЕТИЧНИ ДАННИ
Псевдохипопаратироидизмът (PHP) е генетично хетерогенно заболяване. Данните за вида на наследственото предаване са противоречиви: както Х-свързан доминантен, така и автозомно-доминантен, автозомно-рецесивен тип. В повечето случаи развитието на наследствената остеодистрофия на Олбрайт се свързва с мутации в локуса 20q13 на гена GNAS1, разположен на хромозома 20 (Patten et al., 1990), кодиращ Gs-алфа протеина, свързан с рецептора на паратироидния хормон (PTH). . Подобен фенотип беше открит и при пациенти с интерстициална делеция на дългото рамо на хромозома 2 в локуса 2q37.
ПАТОГЕНЕЗА
Патогенезата на псевдохипопаратиреоидизма се основава на генетично обусловена резистентност на бъбреците и скелета към действието на паратиреоидния хормон в резултат на дефект в комплекса „специфичен циторецептор – паратиреоиден хормон – аденилатциклаза“, който нарушава образуването на цикличен 3"- , 5"-аденозин монофосфат (сАМР) в бъбреците, който е вътреклетъчен медиатор на действието на паратироидния хормон върху метаболитните процеси. Псевдохипопаратиреоидизмът е генетичен хетерогенно заболяване.При някои пациенти самият циторецептор, който свързва паратиреоидния хормон, е дефектен (тип 1А псевдохипопаратиреоидизъм); други имат дефект в нуклеотид-свързващия протеин, локализиран в липидния двоен слой на клетъчната мембрана и функционално свързващ рецептора с аденилат циклазата (тип 1В). псевдохипопаратироидизъм). Някои пациенти изпитват ензимен дефицитсамата аденилат циклаза (псевдохипопаратироидизъм тип 2). Дефицитът на cAMP в резултат на тези дефекти води до нарушаване на синтеза на специфични протеини, които определят биологичния ефект на паратироидния хормон. По този начин се губи чувствителността на целевите органи към паратироидния хормон.
КЛИНИЧНА ХАРАКТЕРИСТИКА
В момента има 4 клинични формипатологии: типове 1A, 1B, 1C и 2. Познаване на техните клинични и биохимични характеристики и данни генетични изследваниядава възможност за диференциална диагноза в рамките на самата нозологична форма.
Честите признаци, които позволяват да се подозира заболяването, са диспропорционалност във физическото развитие, нисък ръст (до степен на нанизъм) поради скъсяване на долните крайници (снимка 1), брахидактилия (снимка 2) и кръгла „лунообразна форма“ лице (снимка 3). Понякога се наблюдават екзостози и зъбна аплазия.
Снимка 1.
Външен вид на дете с остеодистрофия на Олбрайт
(характеристики на фенотипа, нисък ръст поради скъсяване на долните крайници)
Снимка 2.
Характеристики на скелетната система на пациента
с остеодистрофия на Олбрайт
(брахидактилия - скъсяване на пръстите)
Снимка 3.
Характеристики на фенотипа на детето
с остеодистрофия на Олбрайт
(кръгло лице с форма на луна)
Патогномоничен признак е рязкото скъсяване на I, III и V метакарпални и метатарзални кости (особено III и IV), в резултат на което II пръсти на ръцете и краката са по-дълги от останалите и когато ръката е стисната в юмрук, няма изпъкналости в областта на IV и V метакарпофалангеални стави - т. нар. брахиметафалангизъм. Откриват се също къси широки фаланги, удебеляване на черепния свод и деминерализация на костите (остеопороза), затлъстяване.
Умствена изостаналост(обикновено умерена тежест) се открива при приблизително 20% от пациентите. Според някои автори умствената изостаналост се среща в 70% от случаите при наличие на хипокалциемия и в 30% от случаите при нормокалцемия. Психични процесипри пациентите се забавят. В неврологичния статус често се отбелязват двигателна неловкост и невротични реакции: страхове, тревожност, безпокойство, лош сън, повишени рефлекси, конвулсии, които са тетанични по природа и причинени от хипокалцемия, а понякога и конвулсивни пароксизми. Описани са и миопатични симптоми: мускулна умора, мускулна слабост. Често се наблюдават екстрапирамидни нарушения: хореиформна хиперкинеза, атетоза, лицев хемиспазъм, паркинсонизъм; в някои случаи се появяват епилептични пароксизми, церебеларни симптоми: атаксия, загуба на координация.
Често се откриват калцификации на меките тъкани и подкожни калцификации (гърди, корем, сухожилия на петата), като хистологичното им изследване показва остеома кутис(Izraeli et al., 1992), мозък (базални ганглии). Важно е да се отбележи, че калцификатите може вече да са налице при раждането. В резултат на хипокалцемия обикновено се развива катаракта и възникват дефекти на зъбния емайл.
ПСЕВДОХИПОПАРАТРОЗА ТИП 1А
има автозомно доминантен модел на наследяване. Генът за псевдохипопаратиреоидизъм тип 1А - GNAS1 - е локализиран на дългото рамо на хромозома 20, в локуса 20q13.2. Развитието на заболяването е свързано с дефицит на гуанин нуклеотид-свързващ протеин (Gs протеин). В същото време PTH, свързвайки се с рецепторите на целевите тъкани, не е в състояние да активира цикличния аденозин монофосфат (cAMP) и да предизвика тъканен отговор. Вероятно подобен механизъм е в основата на развитието на нечувствителност на тъканите на други органи и ендокринни жлези (хипофункция на щитовидната жлеза, половите жлези, хипофизната жлеза, захарен диабет, както и намалена реакция на черния дроб към приложението на глюкагон), наблюдавана при псевдохипопаратироидизъм тип 1А. При този тип патология не се наблюдава нормалната повишена екскреция на сАМР в урината в отговор на екзогенно приложение на ПТХ. Заболяването се диагностицира по-често на възраст 5-10 години. Болните са с нисък ръст, къс врат, кръгло лице, скъсяване на метакарпалните и метатарзалните кости (обикновено скъсяване на четвъртия и по-рядко втория пръст) - така нареченият брахи-метафалангизъм. Отбелязва се калцификация на меките тъкани и подкожни калцификации, които могат да бъдат открити при раждането; често се наблюдава едновременно засягане на други ендокринни жлези: щитовидна жлеза (хипофункция), гонади, панкреас (захарен диабет). В резултат на хипокалцемия често се развиват катаракта и дефекти на зъбния емайл. Като диференциално диагностичен тест за разграничаване на PHP тип 1A от хипопаратироидизъм: липсата на клиничен ефект от парентералното приложение на ПТХ под формата на повишаване на нивото на калций в кръвта и увеличаване на бъбречната екскреция на фосфор в урината (фосфатен ефект).
Биохимичното изследване разкрива хипокалцемия, хиперфосфатемия, повишени нива на паратироидния хормон в кръвта и хипофосфатурия. Нивото на Gs протеин в кръвта е намалено. Рентгеновото изследване на скелетната система разкрива скъсяване на метакарпалните и метатарзалните кости, генерализирана деминерализация и удебеляване на костите на черепния свод.
ПСЕВДОХИПОПАРАТРОЗА ТИП 1Б
има автозомно доминантен тип наследяване, но не е изключен доминантен, Х-свързан тип наследяване. Необходимо е да се има предвид наблюдаваното понякога непълно проникване на гена на заболяването и възможността за латентно носителство на патологията. Поради това се препоръчва клинично (откриване на субклинично протичане на заболяването) и биохимично изследване (определяне на нивата на калций, фосфор, ПТХ в кръвта) на предполагаеми носители на заболяването. PHP тип 1B се причинява от дефицит на тъканни рецептори за паратиреоиден хормон в прицелните органи и ограничена резистентност към паратиреоиден хормон. Клиничната картина е подобна на тази при тип 1А, но няма увреждане на други ендокринни жлези, по-рядко се среща остеодистрофия.
Пациентите нямат бъбречен отговор на екзогенно приложение на паратиреоиден хормон под формата на повишена екскреция на цикличен аденозин монофосфат в урината; обаче, за разлика от тип 1А, нивото на Gs протеин в кръвта е нормално. Жените са засегнати по-често от мъжете, но тежестта на заболяването може да бъде еднаква както при мъжете, така и при жените.
ПСЕВДОХИПОПАРАТРОЗА ТИП 1C
някои автори го идентифицират с псевдо-псевдохипопаратироидизъм (PPHP), описан от Albright F. през 1952 г. Характеризира се с характерен PGP клинична картинано нивата на калций и фосфор в кръвта и урината остават в нормални граници. Нивата на PTH и Gs протеин в кръвта също остават на нормални нива. Някои пациенти с PHP тип 1C имат делеции de novoна хромозома 2. Възможно е този вариант на заболяването да е подвид на PHP тип 1А.
ПСЕВДОХИПОПАРАТРОЗА ТИП 2
клинично подобен на други видове заболяване, но има автозомно-рецесивен начин на наследяване. Не може да се изключи наличието на автозомно-доминантни форми на патология. Патогенезата на развитието е свързана с вътреклетъчна резистентност към сАМР. След това PTH се свързва с рецепторите и предизвиква нормален клетъчен отговор към PTH под формата на повишена екскреция на cAMP. Вътреклетъчната нечувствителност към сАМР обаче не позволява да се реализира пълният ефект на ПТХ. В същото време нормалната реакция на бъбреците към екзогенно приложение на паратиреоиден хормон се поддържа под формата на повишена екскреция на цикличен аденозин монофосфат в урината. Предполага се, че PHP тип 2 може да бъде свързан с дефицит на витамин D.
По този начин идентифицираните видове PGP се характеризират клинично с намалена чувствителност на целевите органи към ПТХ, но се различават по патогенетичните механизми на образуване на тъканна нечувствителност.
ДИАГНОСТИКА
Лабораторен диференциално диагностичен тест може да бъде моделът на бъбречна екскреция на сАМР в отговор на прилагане на ПТХ: повишена екскреция на сАМР се наблюдава при тип 2 и липсата му при тип 1. Диагнозата се потвърждава от откриването на намалено ниво на гуанин нуклеотид свързващ протеин (Gs протеин) в кръвта (средно 1,5-2 пъти) в сравнение с нормата. Хипокалциемията обикновено се комбинира с хиперфосфатемия и хипофосфатурия. Нивата на ПТХ са повишени; при тип 1C нивото на ПТХ е нормално, което води до името „псевдохипопаратироидизъм“. Рентгеновото изследване на скелетната система разкрива скъсяване на метакарпалните и метатарзалните кости, често генерализирана деминерализация (остеопороза) и удебеляване на костите на черепния свод. Дерматоглифният модел показва изместване на аксиалния палмарен трирадиус.
Критерии за диагностика:
- нисък ръст;
- кръгло лице;
- забавено нервно-психическо развитие;
- скелетни аномалии;
- нисък серумен калций;
- високо ниво на паратхормон в кръвта;
- намалена уринна екскреция на фосфати и сАМР.
ЛЕЧЕНИЕ И ПРОФИЛАКТИКА
Лечението на хипокалцемия се състои в предписване на калциеви добавки в дози, достатъчни за поддържане на нормални концентрации на калций в кръвта. Голямо значениеима терапия с витамин D. Понастоящем се използват активни метаболити на витамин D - оксидевит, 1-алфа-D3, калцитрин и др., в доза 1-2 mcg/ден с положителен резултат (повишени нива на калций в кръвта, намалени прояви на конвулсивен синдром). Тахистин (0,5–1,5 mg/ден) също е ефективен. Това лекарство повишава абсорбцията на калций в червата и по този начин спомага за повишаване на нивото на калций в кръвта. Антиконвулсивната терапия се използва като допълнителна терапия. Лечението няма забележим ефект върху интелектуалното развитие, но заедно с намаляването на симптомите на конвулсивен синдром се наблюдава регресия неврологични прояви(подкорови нарушения, хореиформна хиперкинеза, атетоза и др.). За да се избегне предозиране на препарати с витамин D, е необходимо да се следи концентрацията на калций в кръвта на всеки 3-7 дни през първите 2 седмици от лечението и всеки месец през следващите 2-3 месеца. След достигане на стабилна концентрация на калций в кръвта е достатъчно да се проверява веднъж на всеки 2-3 месеца. Диета с ограничен прием на фосфор спомага за нормализиране на концентрациите на фосфор и калций в кръвта и премахва симптомите на вторичен хиперпаратиреоидизъм. При недостатъчност на други ендокринни жлези се провежда заместителна терапия с подходящи хормони.
Лечението с паратироиден хормон е неефективно. За облекчаване на конвулсивни атаки се прилага интравенозно 10% разтвор на калциев хлорид или калциев глюконат; перорално - 5-10% разтвор на калциев хлорид, 1 супена лъжица 3-4 пъти на ден: калциев глюконат, калциев лактат - до 10 g на ден.
ПРОГНОЗАза цял живот се определя от тежестта на конвулсивния синдром.
ПРЕДОТВРАТЯВАНЕзаболяване се основава на данни от медицинско генетично консултиране.
МЕДИКО-ГЕНЕТИЧНО КОНСУЛТИРАНЕ
При провеждане на медицинско генетично консултиране трябва да се изхожда от автозомно-доминантния тип наследство и високия (50%) риск от повторение на заболяването в семейството с наследствени форми. За да се идентифицира естеството на вида на наследството, е необходимо да се извърши обстоен прегледродители, тъй като синдромът може да се прояви минимално клинични симптоми. Понастоящем е разработена молекулярно-генетична диагностика на болестта и се подобрява чрез типизиране на мутации в гена GNAS1 на хромозома 20. Методите се разработват пренатална диагностиказаболявания като цяло и отделните им видове.
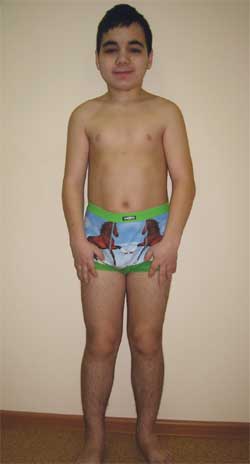
КЛИНИЧНО НАБЛЮДЕНИЕМомче Г., 14,5 години (снимка 4), е прието в Изследователския клиничен институт по педиатрия с диагноза: дегенеративно заболяваненервна система? вродена външна хидроцефалия; симптоматична епилепсия; наследствен синдром? болест на съхранението? метаболитна енцефалопатия; субклиничен хипотиреоидизъм; нисък ръст от смесен произход; когнитивно увреждане.
Оплакванияпри постъпване на интензивно пароксизмално главоболие, локализирано в фронтална области придружено от повръщане, което носи облекчение, намалена памет и успеваемост в училище, гърчове, при което се появяват потрепвания в дясната ръка.
Снимка 4.
Дете Г., 14,5 години, с остеодистрофия на Олбрайт
(фенотипни характеристики, нисък ръст, скъсени крайници, брахидактилия)
Семейна история:родителите са арменци по националност, нямат кръвна връзка и нямат професионални рискове. В родословието на случаите психично заболяване, епилепсия, забавяне на развитието не са отбелязани. Sibs, сестра, на 17 години, се казва, че е здрава.
История на живота и болестта:момче от 2-ра бременност, протекла без особености, второ раждане, на термин, физиологично, тегло - 3100 гр., дължина - 51 см. Извика веднага, оценка по Апгар - 7/9 точки. Влошаване на състоянието на 3-ия ден - неонатални гърчове, спрени в родилния дом. Ранният постнатален период е без особености. Имаше леко забавяне в двигателното развитие през първата година от живота, независимо ходене от 1 година 3 месеца. Във връзка с това е наблюдаван от невролог с диагноза органично увреждане на централната нервна система; вродена хидроцефалия; неонатални гърчове; анамнеза за фебрилни гърчове.
Получих диакарб и финлепсин. Начало на атаките на 1 година и 11 месеца. – асиметрични, тонични под формата на напрежение в дясната ръка и крак, с въртене на очите, до 2 минути, без загуба на съзнание, чести до 10 епизода на ден. Получавах Депакин нередовно. На фона на независимо оттегляне - единичен тонизиращ статус. На 2-годишна възраст е извършена компютърна томография на мозъка по местоживеене, където са идентифицирани единични огнища на демиелинизация в тилните дялове.
Направена е консултация с неврохирург и е препоръчано консервативно лечение. От 3-годишна възраст се наблюдава изоставане в психо-речевото развитие и се препоръчва наблюдение от психиатър.
От 4-5-годишна възраст родителите започват да забелязват деформация и скъсяване на пръстите на ръцете и краката, особено на 2-4 пръста симетрично на ръцете и краката, както и намаляване на показателите за растеж. На 8-годишна възраст заключението на логопеда е общо нарушение на речта от 2-3-то ниво, препоръчва се обучение в специализирано училище. На същата възраст преглед от генетик по местоживеене, заключение: наследствено заболяванеобмен? Препоръчва се изследване на кръвните аминокиселини, не са открити промени; крайно заключение: не са установени данни за наследствено метаболитно заболяване; хипохондроплазия; Препоръчва се лечение при невролог и ендокринолог.
Таблица.
Профил на умственото развитие на дете Г., 14,5 години (IQ = 68)

На 8 години е консултиран от ендокринолог за забавен растеж и развитие. При рентгеново изследване на ръцете се установяват следните особености: средните и основните фаланги и метакарпалните кости са скъсени и удебелени; Диагнозата на рентгенолога е ахондроплазия.
Многократно прегледан по местоживеене в неврологична болница. На 12-годишна възраст се появиха конвулсивни пристъпи без загуба на съзнание с потрепвания на дясната ръка, които бяха серийни, беше назначена антиконвулсивна терапия (депакин), честотата на пристъпите значително намаля. На 13 години е направен ЯМР на мозъка с контраст - симетрични промени в основата на темпоралните дялове на ниво ядра под формата на повишаване на МР сигнала, което е характерно за токсични (манган) или метаболитни (мед, желязо) енцефалопатии.
Прегледана на 13 години 3 месеца. ендокринолог, изследването на профила на щитовидната жлеза разкрива повишаване на тиреостимулиращия хормон (TSH), диагностициран е субклиничен хипотиреоидизъм и е предписан L-тироксин.
При анализиране амбулаторен картондетето и документацията по местоживеене, изследване на калций и фосфор е извършено веднъж, на възраст от 1,5 години, е отбелязана хипокалцемия, но не е извършено допълнително изследване по този въпрос. Като се има предвид несигурността на диагнозата по местоживеене, генетикът изпрати детето в Москва, в Научноизследователския клиничен институт по педиатрия, за да изясни диагнозата.
Данни от обективно изследване:
Височина – 143 см, тегло – 43 кг.
Физическото развитие е много ниско, хармонично, телосложението е непропорционално поради скъсяване на крайниците. Растежът Sds съответства на –2,8 отклонения от нормата (норма –2+2).
Фенотипни характеристики: кръгло лице, къса шия, антимонголоидни палпебрални фисури, широк мост на носа, високо чело, брахидактилия, скъсяване на IV и V метакарпални и метатарзални кости (снимка 5). Вътрешни органи – без особености. Сексуално развитие– Етап по Танер III–IV (което съответства на възрастта).
Лаборатория и функционални изследвания:
Клиничен анализкръвта и урината са нормални.
Биохимичен кръвен тест: общ калций – 1,39 (норма 2,02–2,6 mmol/l), йонизиран калций – 0,61 (норма 1,13–1,32 mmol/l), неорганичен фосфор – 3,66 (норма 0,86–1,56 mmol/l), останалите показатели са в рамките нормални граници.
Биохимичен анализ на урината: бъбречната екскреция на фосфати е намалена - 11,5 mmol/l (норма 19–32 mmol/l).
Профил на щитовидната жлеза: TSH – 11,75 (нормален диапазон 0,4–4,0 µIU/ml), свободен Т4 – 0,49 (нормален диапазон 1,0–1,8 ng/dL).
Паратироиден хормон – 499 (норма 12–65 pg/ml), STH – 7 ng/ml (норма 7–10 ng/ml), соматомедин-С – 250 ng/ml (норма 88–360 ng/ml).
Ехография на вътрешни органи - без особености.
ЕКГ - миграция на суправентрикуларния пейсмейкър на фона на нормална сърдечна честота от 71-80 удара / мин. Непълна блокада десен кракНеговият пакет. Нарушаване на процеса на реполяризация в миокарда задна стеналява камера (намален z.T III, aVF).
R-графия на гръбначния стълб - десностранна сколиоза на гръдния кош 1-ва степен, тежка остеопороза.
R-графия на ръцете с улавяне на предмишниците - скъсяване и разширяване на крайните и средните фаланги. Костна възраст– 13,5–14 години.
Не са регистрирани ЕЕГ модели на епилептична активност.
ЯМР на мозъка - ЯМР картина на множество подкорови огнища на повишен МР сигнал в фронтални дялове, външна компенсирана хидроцефалия с атрофия на мозъчното вещество.
MSCT на мозъка показва симетрични области на калцификация на лентиформени ядра. Дифузни хиперденсни зони в таламуса, опашните ядра с област на калцификация вдясно. Множество точкови калцификации на покривните меки тъкани на черепа.
Аудиограма – без патология.
В ход е ДНК диагностика в гена GNAS1.
Специализирани консултации:
Ендокринолог – Наследствена остеодистрофия на Олбрайт тип 1А (псевдохипопаратироидизъм). Първичен хипотиреоидизъм, непълна лекарствена компенсация.
Офталмолог – пълна вторична катаракта. Препоръчва се хирургично лечение.
Психолог – когнитивно увреждане (психологичният профил на детето е представен в таблицата).
Като се вземат предвид фенотипа на детето, медицинската история, резултатите допълнителни изследвания(хипокалциемия, хиперфосфатемия, хипофосфатурия, повишен паратиреоиден хормон в кръвта), калцификати в мозъка, наличие на катаракта, хипотиреоидизъм), поставена е диагноза: Наследствена остеодистрофия на Олбрайт тип 1А (псевдохипопаратироидизъм). Препоръчва се провеждането на ДНК диагностика - търсене на мутации в гена GNAS1.
Лечение:Детето се препоръчва да приема еутирокс в доза 100 mcg/ден; активен метаболит на витамин D - алфа-D3 (Teva) в доза 2 мкг/ден; калций (Sandoz) 2000 mg/ден; постоянно прилагане на антиконвулсивна терапия - финлепсин 800 mg/ден под наблюдението на невролог-епилептолог; занимания с логопед и психолог; енерготропна терапия (Elkar и коензим Q10 в дози, свързани с възрастта). Проследяване на показателите на фосфорно-калциевия метаболизъм, нивата на паратироидния хормон.
По този начин,Представеното клинично наблюдение показва сложността на диференциално-диагностичното търсене, важността на навременното изследване на прости биохимични параметри (в случай на епилепсия е необходим повторен скрининг на показателите на фосфорно-калциевия метаболизъм), резултатите от късната диагностика на генетично обусловено заболяване , необходимостта от интегриране на отделни признаци в общия фенотип на конкретно патологично състояние за целенасочена навременна диагностика на определени форми на наследствени заболявания. Навременна диагноза, изясняването на генезиса на всеки синдром е особено важно, тъй като ни позволява да намерим оптималния подход към лечението на тези състояния, профилактиката възможни усложнения(до увреждане на детето); предотвратяване на повторна поява на наследствени заболявания в засегнатите семейства (медико-генетично консултиране). Това налага необходимостта лекарите от различни специалности ясно да се ориентират в потока на наследствено обусловената патология. Списъкът с литература е в редакцията.
Синдромът на Олбрайт е наследствена патология с неясен произход. Нарича се още псевдохипопаратироидизъм. Характеризира се с увреждане на костната тъкан. Болестта се проявява предимно в женската половина от населението, въпреки че се среща и при момчетата. Почти всички пациенти имат отклонения в умственото и физическото развитие. Човешките кости започват да се влошават под въздействието на метаболитните процеси на фосфор и калций. Започва отделянето на паратироиден хормон от паращитовидната жлеза.
- Етиология
- Симптоми
- Диагностика
- Лечение
- Предотвратяване
Етиология
Основната причина за такава патология като синдрома на Forbes-Albright е наследствената устойчивост на телесните тъкани към влиянието на паратиреоидния хормон. Ако човек не е болен, тогава хормонът действа чрез определено вещество. Когато заболяването се прояви, съставът на това вещество се променя на генетично ниво. Така периферните тъкани стават нечувствителни към паратироидния хормон.
Симптоми
Синдромът на Олбрайт има три основни симптома:
- Поради високите хормонални нива настъпва ранен пубертет. Момичетата могат да започнат менструация през първите десет месеца от живота си. Но този симптом не се отнася за момчетата. Те започват пубертета както обикновено. Хормоналните нива се повишават, когато надбъбречните жлези, щитовидната жлеза и хипофизата не функционират правилно. Този симптом може да се развие в две форми - непълна и пълна. Ако патологията е пълна, тогава менструацията започва твърде рано. В същото време те са болезнени и изобилни. Появяват се и вторични полови белези, например уголемяване на млечните жлези. Ако формата на патологията е непълна, тогава менструацията не започва. Болестта на Олбрайт при момичетата често е придружена от развитие на кисти на яйчниците. Ранният пубертет при момчетата се характеризира с уголемяване на гениталния орган и поява на пубисно окосмяване.
- Фиброзна остеодисплазия. Това е патология, при която костната тъкан се деформира, което води до огъване на костите на скелета. Проявява се под формата на асиметрия. Визуалните симптоми са гръбначно изкривяване и куцота. Тази патология засяга предимно тръбните кости. Човешкият растеж се забавя. Това може да се забележи дори при дете. Костите на черепа се развиват неравномерно и се появяват изпъкнали очи. След края на пубертета промените в костите спират.
- Промяна в цвета на кожата. Започва да се появява силна пигментация. По бедрата и самото тяло се появяват жълто-кафяви петна с размити ръбове. Петната са ясно видими в долната част на гърба, горната част на гърба, гърдите и задните части. При синдрома на Олбрайт петна се появяват по тялото на детето веднага след раждането.
Заболяването псевдохипопаратироидизъм се проявява със специфични симптоми.
Диагностика
Псевдохипопаратироидизмът се диагностицира въз основа на визуален преглед.
Използват се също лабораторни и хардуерни тестове:
- диагнозата се поставя при поява на два основни симптома;
- назначава се генетична консултация;
- извършва се специална рентгенова снимка;
- извършвам лабораторен анализкръв за хормони;
- необходима е консултация с детски ендокринолог;
- момичетата се преглеждат от детски гинеколог, а момчетата от уролог;
- на момичетата се предписва ултразвуково изследване на органи долната част на таза, а момчетата се подлагат на ултразвук на скротума;
- При необходимост се предписват допълнителни изследвания на ендокринната система.
Пациентите, страдащи от синдрома на Олбрайт, трябва да се подлагат на редовни прегледи от специалисти като ендокринолог, офталмолог, гинеколог и травматолог.
Лечение
Псевдохипопаратироидизмът се лекува комплексно.
Тъй като това е генетична патология, терапията ще бъде насочена към премахване на симптомите, които се появяват:
- Назначава се хормонално лечение. Това ще помогне за функционирането на ендокринната система. Необходимо е внимателно да се следи деформацията на костите на черепа, за да се предотврати развитието на патология навреме.
- Хирургическа интервенция. Този метод се използва, когато деформацията на лицевите кости може да доведе до увреждане на слуха и зрението.
- Предписват се болкоуспокояващи, съдържащи бифосфонати.
За всеки пациент, страдащ от синдрома на Олбрайт, методът на лечение се избира индивидуално. Пациентът трябва стриктно да спазва всички препоръки и предписания на лекаря.
Периодично се проследяват нивата на калций в тялото на пациента. В началния етап на лечението такова изследване се провежда веднъж седмично. Постепенно честотата на измерванията се намалява до веднъж месечно. Ще се провежда до края на назначеното лечение.
Ако се диагностицира псевдохипопаратиреоидизъм, пациентът трябва да се придържа към строга диета. Състои се в пълно премахване от менюто на храни, които съдържат фосфор.
Синдромът на Олбрайт (F. Albright) е заболяване, характеризиращо се с триада от признаци: фиброзна остеодисплазия, огнища на хиперпигментация на кожата и преждевременен пубертет. Много по-често при жените.
Етиологията на синдрома на Олбрайт е неизвестна; предполагат неврогенни влияния и ендокринни промени, причинени от вродени промени в областта на хипоталамуса, с преждевременно производство на гонадотропен хормон на предния дял на хипофизната жлеза поради склероза и хиперостоза на основата на черепа”, главно в областта на sela turcica (фиг. ).
Растежът и зрелостта на скелета (появата и сливането на осификационните ядра) могат значително да надвишат нормалните нива. В резултат на това носителите на синдрома на Олбрайт в детството могат да бъдат по-високи и след преждевременно сливане на епифизите по-ниски от здравите връстници. Напредването на развитието на скелета е придружено от преждевременен пубертет - понякога изтрит, рядко значителен, когато менструалната функция може да се появи през първите години и спорадична менорагия - дори през първите месеци от живота. Преждевременният пубертет е рядък при момчетата.
Огнища на хиперпигментация на кожата при синдрома на Олбрайт се наблюдават на торса, бедрата и по-рядко на шията и краката. Те имат вид на големи полета или отделни и сливащи се петна с диаметър от 2 до 20 cm или повече, с неправилна форма с назъбени ръбове или представени от групи от лунички от бледожълто до кафяво. Понякога тези лезии са видими още при раждането, но ако са малки и бледи, могат да се видят. Локализацията на кожните промени не винаги съвпада с костните лезии. Хиперпигментираните участъци от кожата не се надигат.
При синдрома на Олбрайт се появяват и други аномалии - хипертиреоидизъм, диабет, съдови дефекти, хрущялна остеодисплазия и др. Биохимични показателисъс синдрома на Олбрайт са нормални; понякога активността на алкалната фосфатаза се повишава (до 20 единици на Bodansky). Сред усложненията на фиброзната дисплазия, специфични за синдрома на Олбрайт, са атрофия на зрителния нерв и екзофталмос в резултат на хиперостоза на sela turcica и орбитата.
Прогнозата за синдрома на Олбрайт е благоприятна, но са възможни патологични фрактури и деформация на костите и в редки случаи злокачествено заболяване на фиброзни огнища.
Диференциална диагноза и лечение на костния компонент на синдрома на Олбрайт - вижте Остеодисплазия. Кожните и ендокринните прояви на синдрома на Олбрайт са необратими.
Уплътняване на костната структура на основата на черепа, главно в областта на sela turcica.
Псевдохипопаратироидизъм(синдром на Олбрайт) е заболяване, характеризиращо се с морфологични промени, хипокалциемична тетания и липса на отговор към паратироидния хормон. Заболяването се характеризира с нисък ръст, набито телосложение, кръгло лице, преждевременно затваряне на епифизните осификационни ядра, затлъстяване, брахидактилия, понякога къси метатарзали, дистрофични промени в зъбите и ноктите. Понякога има изоставане в интелигентността и калцификация на меките тъкани. За разлика от истинския хипопаратиреоидизъм, хипокалциемията, хиперфосфатемията и хипофосфатурията не се коригират чрез прилагане на паратиреоиден хормон.
Боледуват обикновено хора: млади и зрели.
Олбрайт обяснява това състояние с рефрактерността на бъбречния тубуларен епител към действието на ендогенен и екзогенен паратироиден хормон, което води до повишена обратна реабсорбция на фосфор от бъбреците с хиперфосфатемия, хипофосфатурия, хиперкалцемия и хиперкалциурия. Той приписва псевдохипопаратиреоидизма на други подобни състояния на органна рефрактерност към определени хормони (виж синдром на Seabright-Bantam).
При това заболяване паращитовидната жлеза има нормална структура.
Псевдопсевдохипопаратироидизъм- патологично състояние, описано през 1952 г. от Олбрайт, напомнящо псевдохипопаратироидизъм с присъщите му морфологични промени, но без съответните хуморални промени.
Нарушения на ендокринната система
Най-често момичетата със синдрома на Олбрайт изпитват преждевременен пубертет, който се причинява от естрогени, освободени в кръвта от киста на яйчника. Кистите могат да се увеличат по размер и след това да намалят в продължение на няколко седмици или дни. С помощта на ултразвукова процедура е възможно да се видят и измерят размерите на туморите. Кистите могат да нараснат до доста прилични размери. Имаше случаи, когато нарасна до размера на топка за голф, тоест повече от 50 мм в диаметър.
Уголемяване на гърдите и менструално кървененаблюдава се заедно с растежа на кистата. Ако едно момиче започне менструация преди 2-годишна възраст, това е първият симптом на синдрома на Олбрайт. Въпреки това, наличието на нередовна менструация и кисти на яйчниците може да се наблюдава както при юноши, така и при възрастни жени. Това не пречи да имате здрави деца.
Лечението на деца с преждевременен пубертет е доста трудно и неефективно. Дори ако кистата се отстрани хирургично, тя може да се появи отново. Приемането на хормона прогестерон може да спре менструацията, но бързата скорост на развитие и растеж на костите няма да се забави. Възможен Отрицателни последициза функционирането на надбъбречните жлези. Лечението използва перорални лекарства, които блокират синтеза на естроген.
Функции на щитовидната жлеза
50 процента от хората със синдром на Олбрайт имат дисфункция на щитовидната жлеза. Това са така наречените гуши, възли и кисти. В редки случаи са възможни леки структурни промени. При тези пациенти нивото на тироид-стимулиращия хормон, произвеждан от хипофизната жлеза, е ниско, но нивата на тиреоидните хормони са нормални или леко повишени. Лечението се провежда за намаляване на синтеза на тиреоидни хормони. Показан е в случаите, когато нивото на секретираните хормони е доста високо.

Прекомерна секреция на растежен хормон
При възникване на заболяването хипофизната жлеза започва да отделя голямо количество растежен хормон. Акромегалия е открита при деца, които са били диагностицирани със синдром на Олбрайт. Младите мъже започнаха да развиват груби черти на лицето, ръцете и краката им бързо се увеличиха и можеха да страдат от артрит. Лечението на деца с такива признаци се свежда до хирургично отстраняване на хипофизната жлеза и използването на синтезирани аналози на хормона соматостатин, които потискат производството на растежен хормон.
Други ендокринни нарушения
Доста рядко налични прекомерна секрецияи уголемяване на надбъбречните жлези. Такова нарушение може да доведе до затлъстяване на тялото и лицето, наддаване на тегло, спиране на растежа и крехкост на кожата. Всички тези симптоми бяха наречени синдром на Кушинг. При такива промени засегнатата надбъбречна жлеза се отстранява или се приемат лекарства, които намаляват синтеза на кортизол.
Понякога децата, които имат синдром на Олбрайт, имат много ниско нивофосфор поради голямата загуба на фосфати в урината. Това разстройство може да е отговорно за промените в костите, свързани с рахит. Лечението включва перорален фосфат и допълнителен витамин D.
Свързани с кожата нарушения
Петната от Cafe-au-lait се появяват по кожата от раждането или малко след това. Най-често се появяват на сакрума, тялото, крайниците, задните части, задна повърхностшията, челото, темето, задната част на главата. Всичко това също са признаци, че детето има синдром на Олбрайт. Снимки на тези места можете да видите по-долу.

Въпреки че при заболяване като неврофиброматоза, има и петна от кафе с мляко. Въпреки това, синдромът на Олбрайт се характеризира с по-големи петна с неправилни очертания и по-малко на брой. Те са с диаметър от 1 до няколко сантиметра и са кафяви на цвят. Всички са с еднакъв цвят, овална форма и гладка повърхност. Хистологичните изследвания най-често показват, че епидермисът не е променен в структурата си, но количеството на меланин в кератиноцитите е леко увеличено.
Единични петна от този тип могат да се появят в доста здрави хора. Ако те не ви притесняват и не растат, тогава няма нужда от лечение. Ако се наблюдава интензивен растеж, има петна с неправилна форма, тогава се препоръчва да се изследват хистологично. И след това го отстранете хирургически.

Заключение
По този начин можем да кажем, че синдромът на Олбрайт се характеризира с увреждане на костите или черепа, наличието на пигментни петна по кожата и ранен пубертет. Въпреки че има случаи, когато са налице само първите два симптома. Като цяло, основният симптом на синдрома са костни лезии (остеодисплазия). С настъпването на пубертета обаче този процес спира. При възрастни костните промени не прогресират. Като цяло, ако се идентифицира и правилно лекува, прогнозата за лечение на това заболяване е доста благоприятна.